 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgnosiophobia in a virtual agent: behavioral and dynamical architecture in Lenia
May 29, 2026All embodied agents are fundamentally patterns in physiological or other excitable media, blurring the distinction between objects and processes. Emergent patterns with complex behaviors, such as Gliders in the Game of Life and virtual patterns in Lenia, are powerful model systems in which to understand the properties and origins of behavioral traits in novel agents. To evaluate the behavior of patterns in Lenia, we introduce regions into their environment from which no sensory information is available - in effect, making creatures blind to parts of their surroundings. Complementing the conventional concept of infotaxis, we find that creatures tend to avoid these regions, a behavior we term agnosiophobia. To explain this behavior, we map each test creature's sensitivity to targeted occlusions and interpret the results in the language of dynamical systems. We observe Lenia creatures taking advantage of their freedom to change heading in order to achieve what appears to be a more fundamental goal: the preservation of their morphology. This work illustrates the beginning of an important roadmap to understand how emergent agents' behavioral propensities interact with the informational, not only tangible, topography of their world.
On learning functions over biological sequence space: relating Gaussian process priors, regularization, and gauge fixing
Apr 26, 2025Mappings from biological sequences (DNA, RNA, protein) to quantitative measures of sequence functionality play an important role in contemporary biology. We are interested in the related tasks of (i) inferring predictive sequence-to-function maps and (ii) decomposing sequence-function maps to elucidate the contributions of individual subsequences. Because each sequence-function map can be written as a weighted sum over subsequences in multiple ways, meaningfully interpreting these weights requires "gauge-fixing," i.e., defining a unique representation for each map. Recent work has established that most existing gauge-fixed representations arise as the unique solutions to $L_2$-regularized regression in an overparameterized "weight space" where the choice of regularizer defines the gauge. Here, we establish the relationship between regularized regression in overparameterized weight space and Gaussian process approaches that operate in "function space," i.e. the space of all real-valued functions on a finite set of sequences. We disentangle how weight space regularizers both impose an implicit prior on the learned function and restrict the optimal weights to a particular gauge. We also show how to construct regularizers that correspond to arbitrary explicit Gaussian process priors combined with a wide variety of gauges. Next, we derive the distribution of gauge-fixed weights implied by the Gaussian process posterior and demonstrate that even for long sequences this distribution can be efficiently computed for product-kernel priors using a kernel trick. Finally, we characterize the implicit function space priors associated with the most common weight space regularizers. Overall, our framework unifies and extends our ability to infer and interpret sequence-function relationships.
Inferring genotype-phenotype maps using attention models
Apr 14, 2025Predicting phenotype from genotype is a central challenge in genetics. Traditional approaches in quantitative genetics typically analyze this problem using methods based on linear regression. These methods generally assume that the genetic architecture of complex traits can be parameterized in terms of an additive model, where the effects of loci are independent, plus (in some cases) pairwise epistatic interactions between loci. However, these models struggle to analyze more complex patterns of epistasis or subtle gene-environment interactions. Recent advances in machine learning, particularly attention-based models, offer a promising alternative. Initially developed for natural language processing, attention-based models excel at capturing context-dependent interactions and have shown exceptional performance in predicting protein structure and function. Here, we apply attention-based models to quantitative genetics. We analyze the performance of this attention-based approach in predicting phenotype from genotype using simulated data across a range of models with increasing epistatic complexity, and using experimental data from a recent quantitative trait locus mapping study in budding yeast. We find that our model demonstrates superior out-of-sample predictions in epistatic regimes compared to standard methods. We also explore a more general multi-environment attention-based model to jointly analyze genotype-phenotype maps across multiple environments and show that such architectures can be used for "transfer learning" - predicting phenotypes in novel environments with limited training data.
Cortical Computation via Iterative Constructions
Jun 15, 2016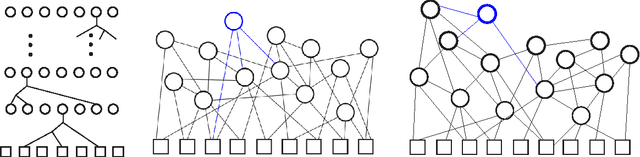



We study Boolean functions of an arbitrary number of input variables that can be realized by simple iterative constructions based on constant-size primitives. This restricted type of construction needs little global coordination or control and thus is a candidate for neurally feasible computation. Valiant's construction of a majority function can be realized in this manner and, as we show, can be generalized to any uniform threshold function. We study the rate of convergence, finding that while linear convergence to the correct function can be achieved for any threshold using a fixed set of primitives, for quadratic convergence, the size of the primitives must grow as the threshold approaches 0 or 1. We also study finite realizations of this process and the learnability of the functions realized. We show that the constructions realized are accurate outside a small interval near the target threshold, where the size of the construction grows as the inverse square of the interval width. This phenomenon, that errors are higher closer to thresholds (and thresholds closer to the boundary are harder to represent), is a well-known cognitive finding.