 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLLM-Based Evaluation of Low-Resource Machine Translation: A Reference-less Dialect Guided Approach with a Refined Sylheti-English Benchmark
May 18, 2025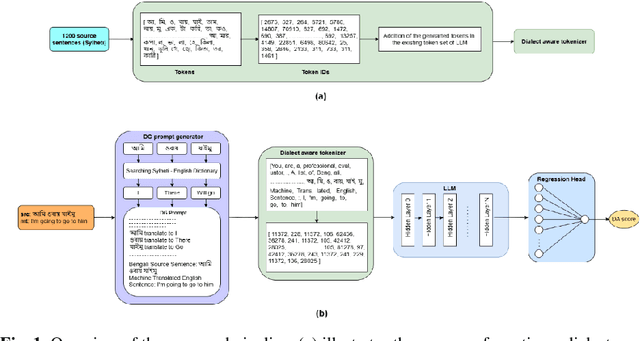
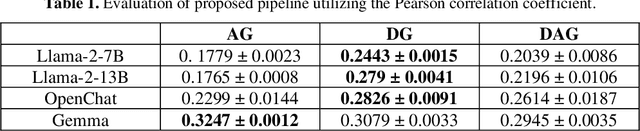

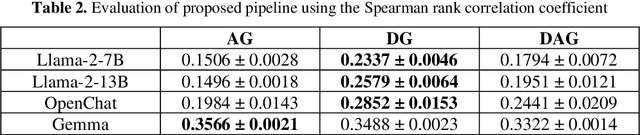
Evaluating machine translation (MT) for low-resource languages poses a persistent challenge, primarily due to the limited availability of high quality reference translations. This issue is further exacerbated in languages with multiple dialects, where linguistic diversity and data scarcity hinder robust evaluation. Large Language Models (LLMs) present a promising solution through reference-free evaluation techniques; however, their effectiveness diminishes in the absence of dialect-specific context and tailored guidance. In this work, we propose a comprehensive framework that enhances LLM-based MT evaluation using a dialect guided approach. We extend the ONUBAD dataset by incorporating Sylheti-English sentence pairs, corresponding machine translations, and Direct Assessment (DA) scores annotated by native speakers. To address the vocabulary gap, we augment the tokenizer vocabulary with dialect-specific terms. We further introduce a regression head to enable scalar score prediction and design a dialect-guided (DG) prompting strategy. Our evaluation across multiple LLMs shows that the proposed pipeline consistently outperforms existing methods, achieving the highest gain of +0.1083 in Spearman correlation, along with improvements across other evaluation settings. The dataset and the code are available at https://github.com/180041123-Atiq/MTEonLowResourceLanguage.
A Pan-cancer Classification Model using Multi-view Feature Selection Method and Ensemble Classifier
Jan 12, 2025



Accurately identifying cancer samples is crucial for precise diagnosis and effective patient treatment. Traditional methods falter with high-dimensional and high feature-to-sample count ratios, which are critical for classifying cancer samples. This study aims to develop a novel feature selection framework specifically for transcriptome data and propose two ensemble classifiers. For feature selection, we partition the transcriptome dataset vertically based on feature types. Then apply the Boruta feature selection process on each of the partitions, combine the results, and apply Boruta again on the combined result. We repeat the process with different parameters of Boruta and prepare the final feature set. Finally, we constructed two ensemble ML models based on LR, SVM and XGBoost classifiers with max voting and averaging probability approach. We used 10-fold cross-validation to ensure robust and reliable classification performance. With 97.11\% accuracy and 0.9996 AUC value, our approach performs better compared to existing state-of-the-art methods to classify 33 types of cancers. A set of 12 types of cancer is traditionally challenging to differentiate between each other due to their similarity in tissue of origin. Our method accurately identifies over 90\% of samples from these 12 types of cancers, which outperforms all known methods presented in existing literature. The gene set enrichment analysis reveals that our framework's selected features have enriched the pathways highly related to cancers. This study develops a feature selection framework to select features highly related to cancer development and leads to identifying different types of cancer samples with higher accuracy.
Precision Cancer Classification and Biomarker Identification from mRNA Gene Expression via Dimensionality Reduction and Explainable AI
Oct 08, 2024Gene expression analysis is a critical method for cancer classification, enabling precise diagnoses through the identification of unique molecular signatures associated with various tumors. Identifying cancer-specific genes from gene expression values enables a more tailored and personalized treatment approach. However, the high dimensionality of mRNA gene expression data poses challenges for analysis and data extraction. This research presents a comprehensive pipeline designed to accurately identify 33 distinct cancer types and their corresponding gene sets. It incorporates a combination of normalization and feature selection techniques to reduce dataset dimensionality effectively while ensuring high performance. Notably, our pipeline successfully identifies a substantial number of cancer-specific genes using a reduced feature set of just 500, in contrast to using the full dataset comprising 19,238 features. By employing an ensemble approach that combines three top-performing classifiers, a classification accuracy of 96.61% was achieved. Furthermore, we leverage Explainable AI to elucidate the biological significance of the identified cancer-specific genes, employing Differential Gene Expression (DGE) analysis.