 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Comparison of Polynomial-Based Tree Clustering Methods
Jan 13, 2026Tree structures appear in many fields of the life sciences, including phylogenetics, developmental biology and nucleic acid structures. Trees can be used to represent RNA secondary structures, which directly relate to the function of non-coding RNAs. Recent developments in sequencing technology and artificial intelligence have yielded numerous biological data that can be represented with tree structures. This requires novel methods for tree structure data analytics. Tree polynomials provide a computationally efficient, interpretable and comprehensive way to encode tree structures as matrices, which are compatible with most data analytics tools. Machine learning methods based on the Canberra distance between tree polynomials have been introduced to analyze phylogenies and nucleic acid structures. In this paper, we compare the performance of different distances in tree clustering methods based on a tree distinguishing polynomial. We also implement two basic autoencoder models for clustering trees using the polynomial. We find that the distance based methods with entry-level normalized distances have the highest clustering accuracy among the compared methods.
Graph Based Analysis for Gene Segment Organization In a Scrambled Genome
Jan 28, 2018

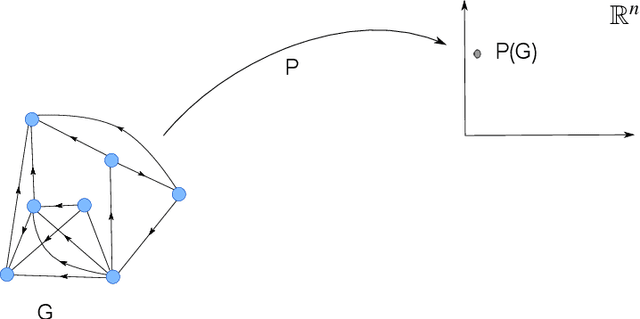
DNA rearrangement processes recombine gene segments that are organized on the chromosome in a variety of ways. The segments can overlap, interleave or one may be a subsegment of another. We use directed graphs to represent segment organizations on a given locus where contigs containing rearranged segments represent vertices and the edges correspond to the segment relationships. Using graph properties we associate a point in a higher dimensional Euclidean space to each graph such that cluster formations and analysis can be performed with methods from topological data analysis. The method is applied to a recently sequenced model organism \textit{Oxytricha trifallax}, a species of ciliate with highly scrambled genome that undergoes massive rearrangement process after conjugation. The analysis shows some emerging star-like graph structures indicating that segments of a single gene can interleave, or even contain all of the segments from fifteen or more other genes in between its segments. We also observe that as many as six genes can have their segments mutually interleaving or overlapping.