 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAnnotateMissense: a genome-wide annotation and benchmarking framework for missense pathogenicity prediction
May 23, 2026Missense variant interpretation remains challenging because pathogenicity depends on heterogeneous evidence from population frequency, evolutionary conservation, transcript context, amino acid substitution severity, prior pathogenicity predictors and protein-language-model-derived features. We present AnnotateMissense, a scalable annotation, benchmarking and genome-wide prediction framework for missense variant interpretation. AnnotateMissense integrates hg38 missense variants derived from dbNSFP v5.1 with ANNOVAR annotations, dbNSFP transcript/protein descriptors, AlphaMissense scores, ESM-derived features, conservation metrics, population-frequency variables, established pathogenicity predictors and engineered amino acid/codon-context features. Using 132,714 ClinVar-labelled missense variants, we benchmarked machine-learning and deep-learning models under controlled feature configurations. The full 303-feature benchmark set achieved the strongest performance with XGBoost, reaching mean MCC = 0.9411 and ROC-AUC = 0.9950 across stratified five-fold cross-validation. Restricted naive and location-oriented feature sets achieved lower best MCC values of 0.4989 and 0.5113, respectively. Circularity-controlled ablations showed that removing prior-predictor, population-frequency and clinically overlapping evidence reduced performance, whereas excluding AlphaMissense and ESM-derived features alone had minimal effect. Temporal ClinVar validation on newly observed pathogenic/benign variants achieved MCC = 0.7613, accuracy = 0.8798 and F1-score = 0.8750. The final model was applied to 90,643,830 hg38 missense variants to generate AnnotateMissense pathogenicity scores and binary prediction labels. Code and outputs are available at https://github.com/MuhammadMuneeb007/CAGI7_Annotate_All_Missense and https://doi.org/10.5281/zenodo.19981867.
G2DR: A Genotype-First Framework for Genetics-Informed Target Prioritization and Drug Repurposing
Mar 20, 2026Human genetics offers a promising route to therapeutic discovery, yet practical frameworks translating genotype-derived signal into ranked target and drug hypotheses remain limited, particularly when matched disease transcriptomics are unavailable. Here we present G2DR, a genotype-first prioritization framework propagating inherited variation through genetically predicted expression, multi-method gene-level testing, pathway enrichment, network context, druggability, and multi-source drug--target evidence integration. In a migraine case study with 733 UK Biobank participants under stratified five-fold cross-validation, we imputed expression across seven transcriptome-weight resources and ranked genes using a reproducibility-aware discovery score from training and validation data, followed by a balanced integrated score for target selection. Discovery-based prioritization generalized to held-out data, achieving gene-level ROC-AUC of 0.775 and PR-AUC of 0.475, while retaining enrichment for curated migraine biology. Mapping prioritized genes to compounds via Open Targets, DGIdb, and ChEMBL yielded drug sets enriched for migraine-linked compounds relative to a global background, though recovery favoured broader mechanism-linked and off-label space over migraine-specific approved therapies. Directionality filtering separated broadly recovered compounds from mechanistically compatible candidates. G2DR is a modular framework for genetics-informed hypothesis generation, not a clinically actionable recommendation system. All outputs require independent experimental, pharmacological, and clinical validation.
Deep learning pipeline for image classification on mobile phones
May 31, 2022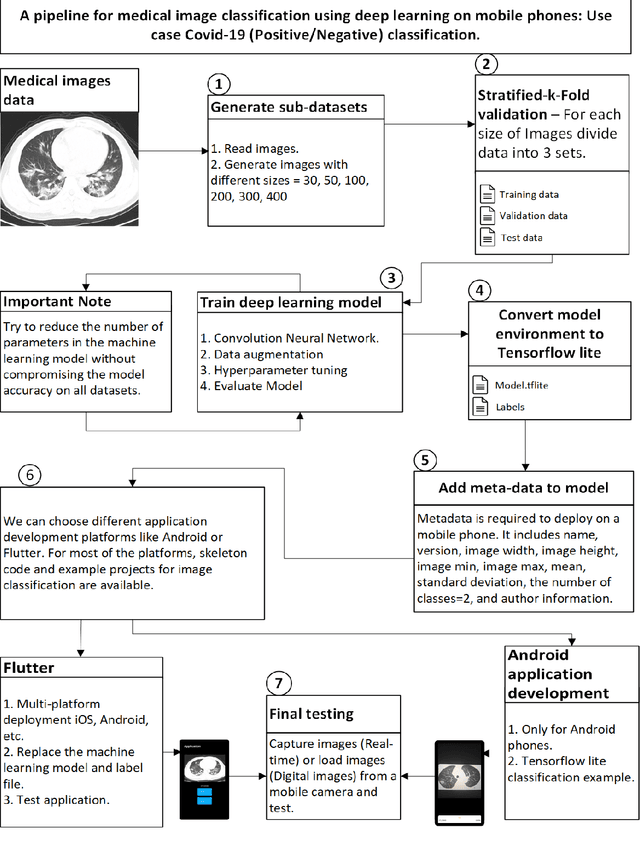
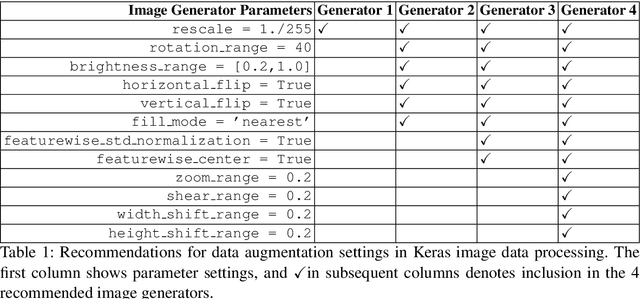
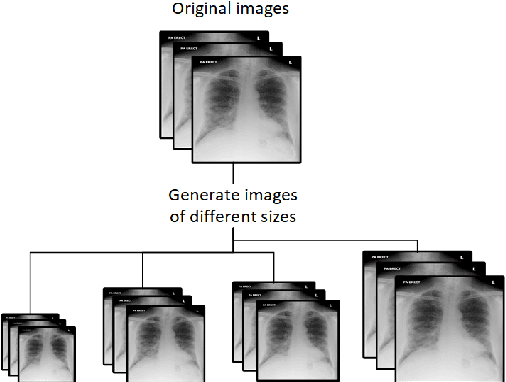
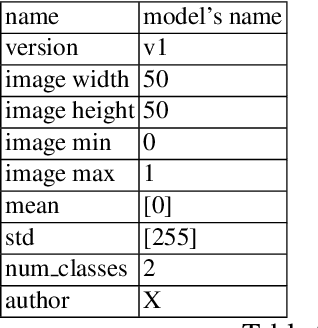
This article proposes and documents a machine-learning framework and tutorial for classifying images using mobile phones. Compared to computers, the performance of deep learning model performance degrades when deployed on a mobile phone and requires a systematic approach to find a model that performs optimally on both computers and mobile phones. By following the proposed pipeline, which consists of various computational tools, simple procedural recipes, and technical considerations, one can bring the power of deep learning medical image classification to mobile devices, potentially unlocking new domains of applications. The pipeline is demonstrated on four different publicly available datasets: COVID X-rays, COVID CT scans, leaves, and colorectal cancer. We used two application development frameworks: TensorFlow Lite (real-time testing) and Flutter (digital image testing) to test the proposed pipeline. We found that transferring deep learning models to a mobile phone is limited by hardware and classification accuracy drops. To address this issue, we proposed this pipeline to find an optimized model for mobile phones. Finally, we discuss additional applications and computational concerns related to deploying deep-learning models on phones, including real-time analysis and image preprocessing. We believe the associated documentation and code can help physicians and medical experts develop medical image classification applications for distribution.
* 20 pages