 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOmics-scale polymer computational database transferable to real-world artificial intelligence applications
Nov 07, 2025Developing large-scale foundational datasets is a critical milestone in advancing artificial intelligence (AI)-driven scientific innovation. However, unlike AI-mature fields such as natural language processing, materials science, particularly polymer research, has significantly lagged in developing extensive open datasets. This lag is primarily due to the high costs of polymer synthesis and property measurements, along with the vastness and complexity of the chemical space. This study presents PolyOmics, an omics-scale computational database generated through fully automated molecular dynamics simulation pipelines that provide diverse physical properties for over $10^5$ polymeric materials. The PolyOmics database is collaboratively developed by approximately 260 researchers from 48 institutions to bridge the gap between academia and industry. Machine learning models pretrained on PolyOmics can be efficiently fine-tuned for a wide range of real-world downstream tasks, even when only limited experimental data are available. Notably, the generalisation capability of these simulation-to-real transfer models improve significantly as the size of the PolyOmics database increases, exhibiting power-law scaling. The emergence of scaling laws supports the "more is better" principle, highlighting the significance of ultralarge-scale computational materials data for improving real-world prediction performance. This unprecedented omics-scale database reveals vast unexplored regions of polymer materials, providing a foundation for AI-driven polymer science.
A Bayesian algorithm for retrosynthesis
Mar 06, 2020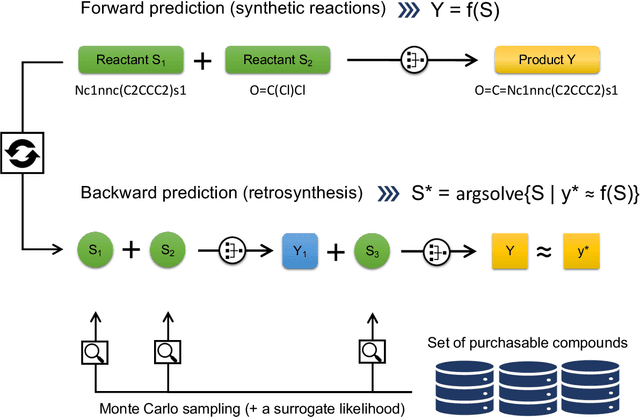
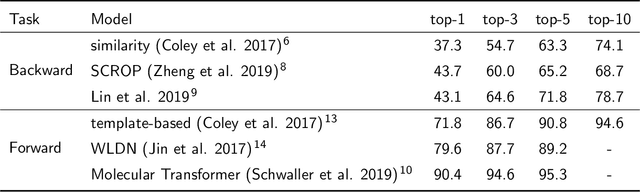
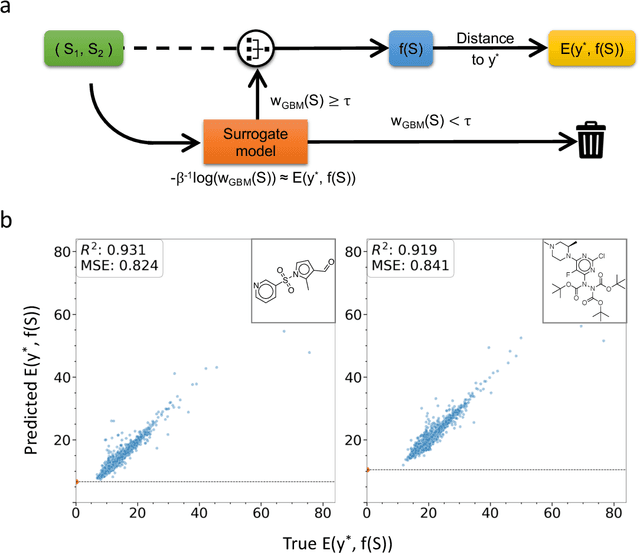

The identification of synthetic routes that end with a desired product has been an inherently time-consuming process that is largely dependent on expert knowledge regarding a limited fraction of the entire reaction space. At present, emerging machine-learning technologies are overturning the process of retrosynthetic planning. The objective of this study is to discover synthetic routes backwardly from a given desired molecule to commercially available compounds. The problem is reduced to a combinatorial optimization task with the solution space subject to the combinatorial complexity of all possible pairs of purchasable reactants. We address this issue within the framework of Bayesian inference and computation. The workflow consists of two steps: a deep neural network is trained that forwardly predicts a product of the given reactants with a high level of accuracy, following which this forward model is inverted into the backward one via Bayes' law of conditional probability. Using the backward model, a diverse set of highly probable reaction sequences ending with a given synthetic target is exhaustively explored using a Monte Carlo search algorithm. The Bayesian retrosynthesis algorithm could successfully rediscover 80.3% and 50.0% of known synthetic routes of single-step and two-step reactions within top-10 accuracy, respectively, thereby outperforming state-of-the-art algorithms in terms of the overall accuracy. Remarkably, the Monte Carlo method, which was specifically designed for the presence of diverse multiple routes, often revealed a ranked list of hundreds of reaction routes to the same synthetic target. We investigated the potential applicability of such diverse candidates based on expert knowledge from synthetic organic chemistry.