 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNavigation Agents for the Visually Impaired: A Sidewalk Simulator and Experiments
Oct 29, 2019
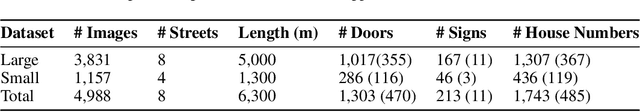

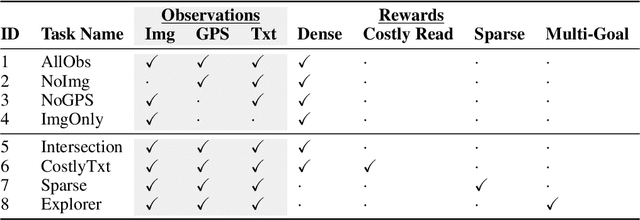
Millions of blind and visually-impaired (BVI) people navigate urban environments every day, using smartphones for high-level path-planning and white canes or guide dogs for local information. However, many BVI people still struggle to travel to new places. In our endeavor to create a navigation assistant for the BVI, we found that existing Reinforcement Learning (RL) environments were unsuitable for the task. This work introduces SEVN, a sidewalk simulation environment and a neural network-based approach to creating a navigation agent. SEVN contains panoramic images with labels for house numbers, doors, and street name signs, and formulations for several navigation tasks. We study the performance of an RL algorithm (PPO) in this setting. Our policy model fuses multi-modal observations in the form of variable resolution images, visible text, and simulated GPS data to navigate to a goal door. We hope that this dataset, simulator, and experimental results will provide a foundation for further research into the creation of agents that can assist members of the BVI community with outdoor navigation.
The TCGA Meta-Dataset Clinical Benchmark
Oct 18, 2019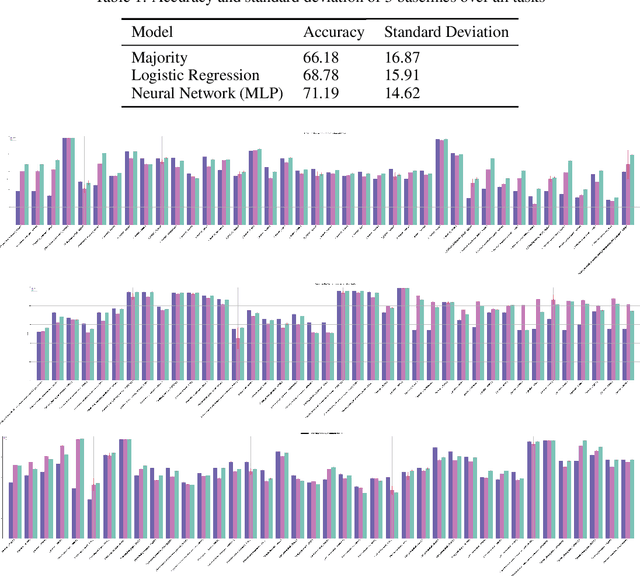
Machine learning is bringing a paradigm shift to healthcare by changing the process of disease diagnosis and prognosis in clinics and hospitals. This development equips doctors and medical staff with tools to evaluate their hypotheses and hence make more precise decisions. Although most current research in the literature seeks to develop techniques and methods for predicting one particular clinical outcome, this approach is far from the reality of clinical decision making in which you have to consider several factors simultaneously. In addition, it is difficult to follow the recent progress concretely as there is a lack of consistency in benchmark datasets and task definitions in the field of Genomics. To address the aforementioned issues, we provide a clinical Meta-Dataset derived from the publicly available data hub called The Cancer Genome Atlas Program (TCGA) that contains 174 tasks. We believe those tasks could be good proxy tasks to develop methods which can work on a few samples of gene expression data. Also, learning to predict multiple clinical variables using gene-expression data is an important task due to the variety of phenotypes in clinical problems and lack of samples for some of the rare variables. The defined tasks cover a wide range of clinical problems including predicting tumor tissue site, white cell count, histological type, family history of cancer, gender, and many others which we explain later in the paper. Each task represents an independent dataset. We use regression and neural network baselines for all the tasks using only 150 samples and compare their performance.
Analysis of Gene Interaction Graphs for Biasing Machine Learning Models
May 06, 2019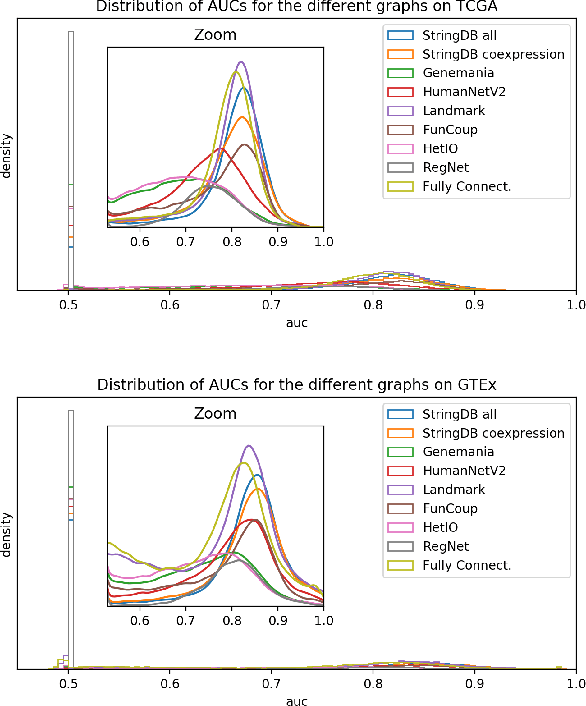
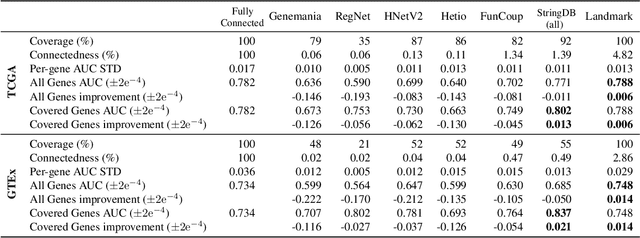
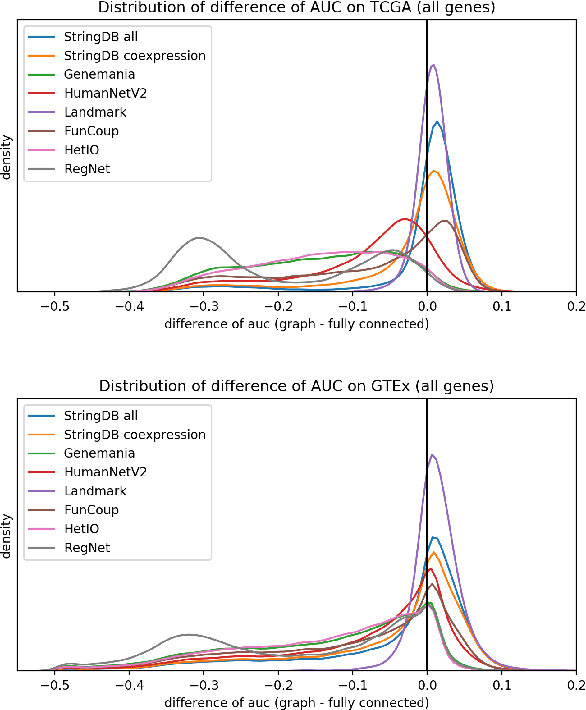
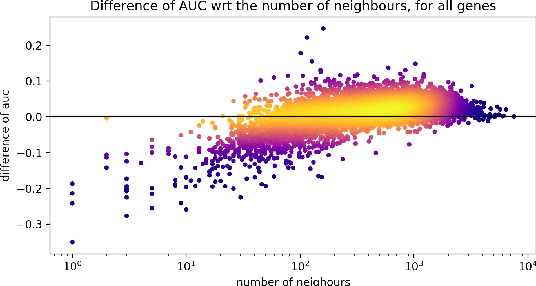
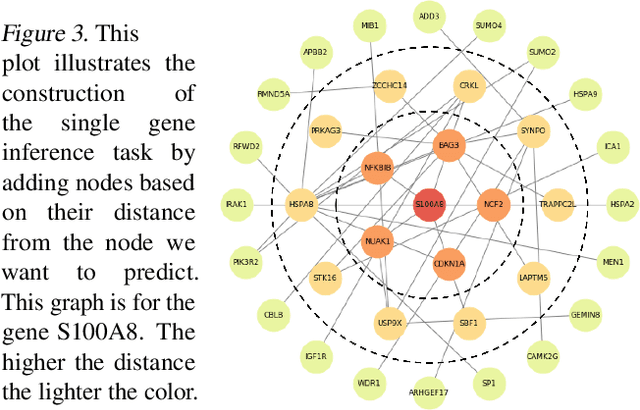
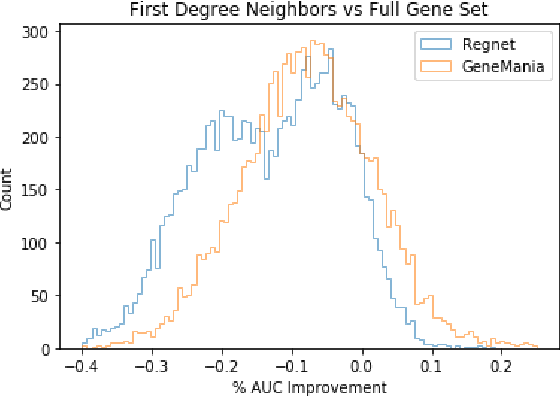
Gene interaction graphs aim to capture various relationships between genes and can be used to create more biologically-intuitive models for machine learning. There are many such graphs available which can differ in the number of genes and edges covered. In this work, we attempt to evaluate the biases provided by those graphs through utilizing them for 'Single Gene Inference' (SGI) which serves as, what we believe is, a proxy for more relevant prediction tasks. The SGI task assesses how well a gene's neighbors in a particular graph can 'explain' the gene itself in comparison to the baseline of using all the genes in the dataset. We evaluate seven major gene interaction graphs created by different research groups on two distinct datasets, TCGA and GTEx. We find that some graphs perform on par with the unbiased baseline for most genes with a significantly smaller feature set.
A Survey of Mobile Computing for the Visually Impaired
Nov 27, 2018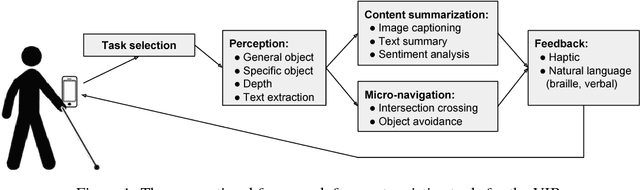
The number of visually impaired or blind (VIB) people in the world is estimated at several hundred million. Based on a series of interviews with the VIB and developers of assistive technology, this paper provides a survey of machine-learning based mobile applications and identifies the most relevant applications. We discuss the functionality of these apps, how they align with the needs and requirements of the VIB users, and how they can be improved with techniques such as federated learning and model compression. As a result of this study we identify promising future directions of research in mobile perception, micro-navigation, and content-summarization.
Towards Gene Expression Convolutions using Gene Interaction Graphs
Jun 18, 2018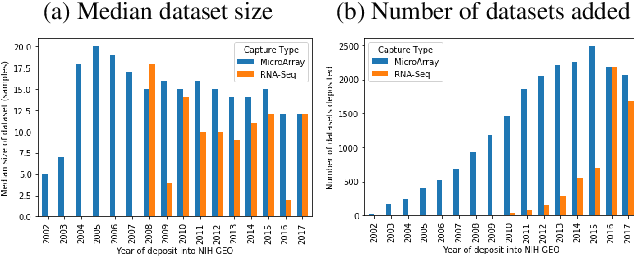
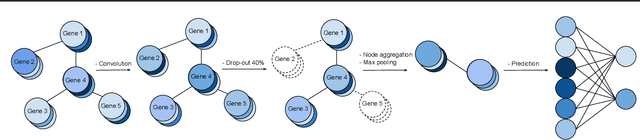
We study the challenges of applying deep learning to gene expression data. We find experimentally that there exists non-linear signal in the data, however is it not discovered automatically given the noise and low numbers of samples used in most research. We discuss how gene interaction graphs (same pathway, protein-protein, co-expression, or research paper text association) can be used to impose a bias on a deep model similar to the spatial bias imposed by convolutions on an image. We explore the usage of Graph Convolutional Neural Networks coupled with dropout and gene embeddings to utilize the graph information. We find this approach provides an advantage for particular tasks in a low data regime but is very dependent on the quality of the graph used. We conclude that more work should be done in this direction. We design experiments that show why existing methods fail to capture signal that is present in the data when features are added which clearly isolates the problem that needs to be addressed.