 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePerspective: Towards sustainable exploration of chemical spaces with machine learning
Mar 31, 2026Artificial intelligence is transforming molecular and materials science, but its growing computational and data demands raise critical sustainability challenges. In this Perspective, we examine resource considerations across the AI-driven discovery pipeline--from quantum-mechanical (QM) data generation and model training to automated, self-driving research workflows--building on discussions from the ``SusML workshop: Towards sustainable exploration of chemical spaces with machine learning'' held in Dresden, Germany. In this context, the availability of large quantum datasets has enabled rigorous benchmarking and rapid methodological progress, while also incurring substantial energy and infrastructure costs. We highlight emerging strategies to enhance efficiency, including general-purpose machine learning (ML) models, multi-fidelity approaches, model distillation, and active learning. Moreover, incorporating physics-based constraints within hierarchical workflows, where fast ML surrogates are applied broadly and high-accuracy QM methods are used selectively, can further optimize resource use without compromising reliability. Equally important is bridging the gap between idealized computational predictions and real-world conditions by accounting for synthesizability and multi-objective design criteria, which is essential for practical impact. Finally, we argue that sustainable progress will rely on open data and models, reusable workflows, and domain-specific AI systems that maximize scientific value per unit of computation, enabling efficient and responsible discovery of technological materials and therapeutics.
Electronic-structure properties from atom-centered predictions of the electron density
Jun 28, 2022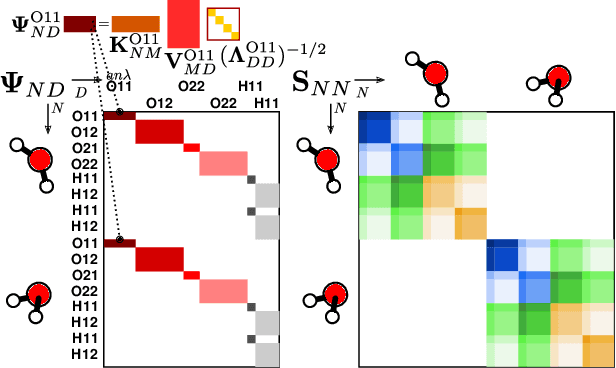


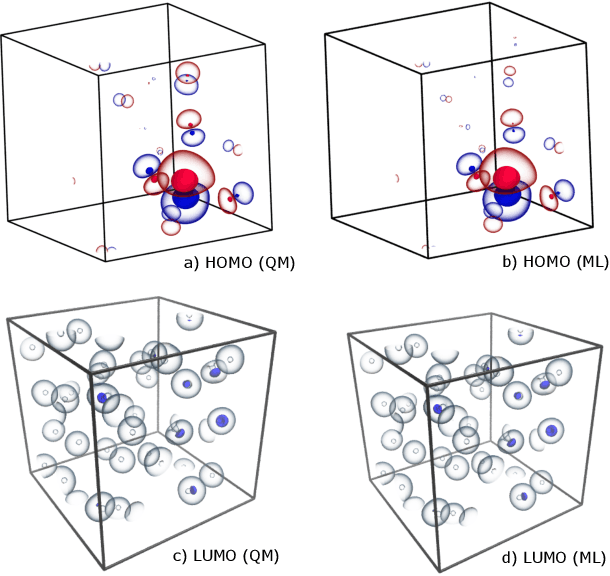
The electron density of a molecule or material has recently received major attention as a target quantity of machine-learning models. A natural choice to construct a model that yields transferable and linear-scaling predictions is to represent the scalar field using a multi-centered atomic basis analogous to that routinely used in density fitting approximations. However, the non-orthogonality of the basis poses challenges for the learning exercise, as it requires accounting for all the atomic density components at once. We devise a gradient-based approach to directly minimize the loss function of the regression problem in an optimized and highly sparse feature space. In so doing, we overcome the limitations associated with adopting an atom-centered model to learn the electron density over arbitrarily complex datasets, obtaining extremely accurate predictions. The enhanced framework is tested on 32-molecule periodic cells of liquid water, presenting enough complexity to require an optimal balance between accuracy and computational efficiency. We show that starting from the predicted density a single Kohn-Sham diagonalization step can be performed to access total energy components that carry an error of just 0.1 meV/atom with respect to the reference density functional calculations. Finally, we test our method on the highly heterogeneous QM9 benchmark dataset, showing that a small fraction of the training data is enough to derive ground-state total energies within chemical accuracy.