 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomatic and explainable grading of meningiomas from histopathology images
Jul 19, 2021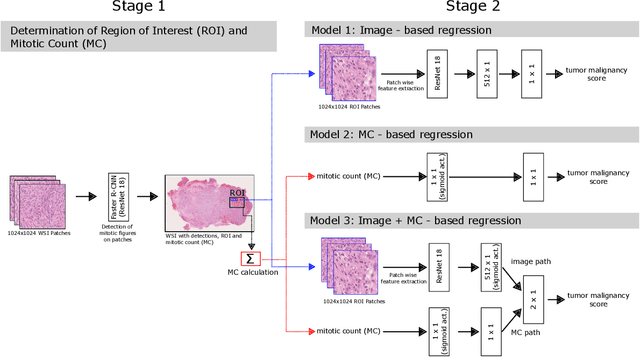
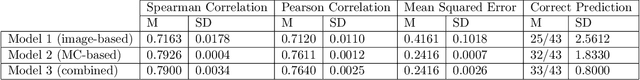
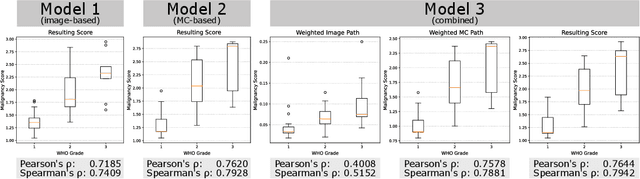
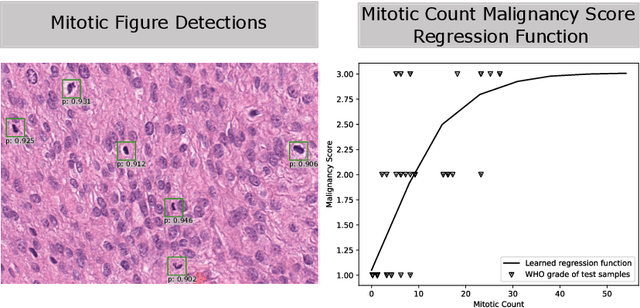
Meningioma is one of the most prevalent brain tumors in adults. To determine its malignancy, it is graded by a pathologist into three grades according to WHO standards. This grade plays a decisive role in treatment, and yet may be subject to inter-rater discordance. In this work, we present and compare three approaches towards fully automatic meningioma grading from histology whole slide images. All approaches are following a two-stage paradigm, where we first identify a region of interest based on the detection of mitotic figures in the slide using a state-of-the-art object detection deep learning network. This region of highest mitotic rate is considered characteristic for biological tumor behavior. In the second stage, we calculate a score corresponding to tumor malignancy based on information contained in this region using three different settings. In a first approach, image patches are sampled from this region and regression is based on morphological features encoded by a ResNet-based network. We compare this to learning a logistic regression from the determined mitotic count, an approach which is easily traceable and explainable. Lastly, we combine both approaches in a single network. We trained the pipeline on 951 slides from 341 patients and evaluated them on a separate set of 141 slides from 43 patients. All approaches yield a high correlation to the WHO grade. The logistic regression and the combined approach had the best results in our experiments, yielding correct predictions in 32 and 33 of all cases, respectively, with the image-based approach only predicting 25 cases correctly. Spearman's correlation was 0.716, 0.792 and 0.790 respectively. It may seem counterintuitive at first that morphological features provided by image patches do not improve model performance. Yet, this mirrors the criteria of the grading scheme, where mitotic count is the only unequivocal parameter.
Quantifying the Scanner-Induced Domain Gap in Mitosis Detection
Mar 30, 2021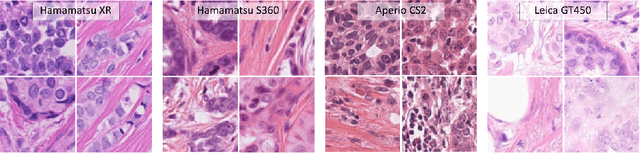

Automated detection of mitotic figures in histopathology images has seen vast improvements, thanks to modern deep learning-based pipelines. Application of these methods, however, is in practice limited by strong variability of images between labs. This results in a domain shift of the images, which causes a performance drop of the models. Hypothesizing that the scanner device plays a decisive role in this effect, we evaluated the susceptibility of a standard mitosis detection approach to the domain shift introduced by using a different whole slide scanner. Our work is based on the MICCAI-MIDOG challenge 2021 data set, which includes 200 tumor cases of human breast cancer and four scanners. Our work indicates that the domain shift induced not by biochemical variability but purely by the choice of acquisition device is underestimated so far. Models trained on images of the same scanner yielded an average F1 score of 0.683, while models trained on a single other scanner only yielded an average F1 score of 0.325. Training on another multi-domain mitosis dataset led to mean F1 scores of 0.52. We found this not to be reflected by domain-shifts measured as proxy A distance-derived metric.
Learning to be EXACT, Cell Detection for Asthma on Partially Annotated Whole Slide Images
Jan 13, 2021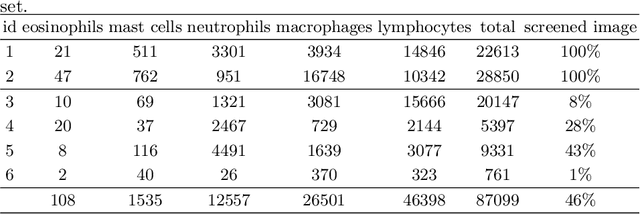
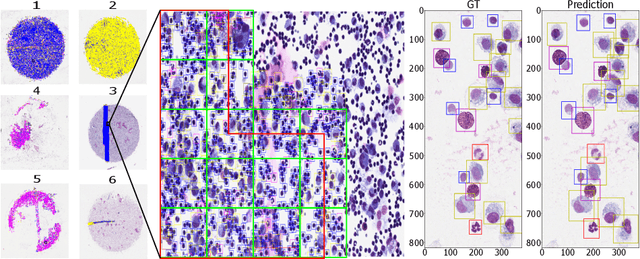
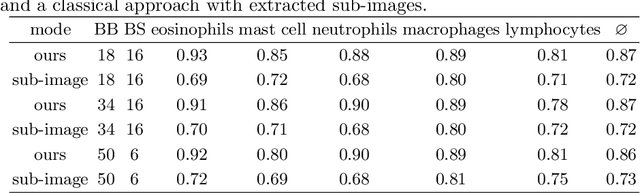
Asthma is a chronic inflammatory disorder of the lower respiratory tract and naturally occurs in humans and animals including horses. The annotation of an asthma microscopy whole slide image (WSI) is an extremely labour-intensive task due to the hundreds of thousands of cells per WSI. To overcome the limitation of annotating WSI incompletely, we developed a training pipeline which can train a deep learning-based object detection model with partially annotated WSIs and compensate class imbalances on the fly. With this approach we can freely sample from annotated WSIs areas and are not restricted to fully annotated extracted sub-images of the WSI as with classical approaches. We evaluated our pipeline in a cross-validation setup with a fixed training set using a dataset of six equine WSIs of which four are partially annotated and used for training, and two fully annotated WSI are used for validation and testing. Our WSI-based training approach outperformed classical sub-image-based training methods by up to 15\% $mAP$ and yielded human-like performance when compared to the annotations of ten trained pathologists.
How Many Annotators Do We Need? -- A Study on the Influence of Inter-Observer Variability on the Reliability of Automatic Mitotic Figure Assessment
Jan 08, 2021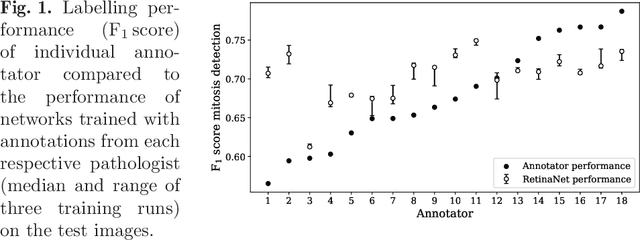
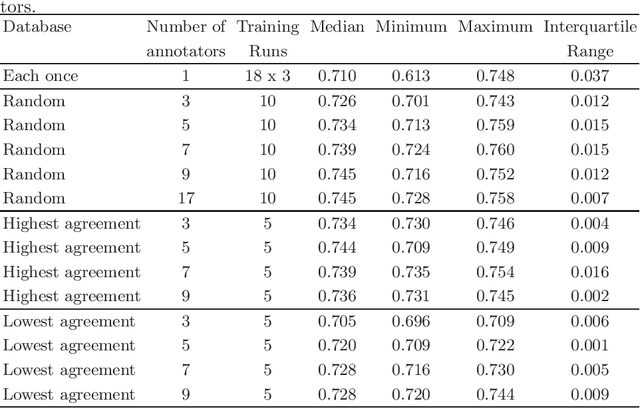
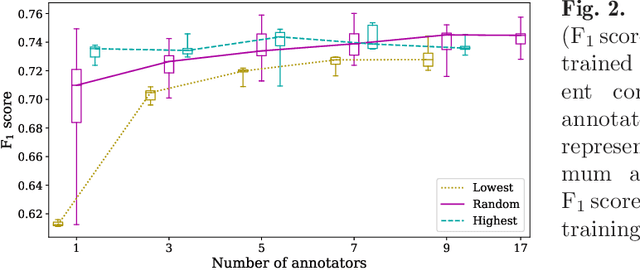
Density of mitotic figures in histologic sections is a prognostically relevant characteristic for many tumours. Due to high inter-pathologist variability, deep learning-based algorithms are a promising solution to improve tumour prognostication. Pathologists are the gold standard for database development, however, labelling errors may hamper development of accurate algorithms. In the present work we evaluated the benefit of multi-expert consensus (n = 3, 5, 7, 9, 11) on algorithmic performance. While training with individual databases resulted in highly variable F$_1$ scores, performance was notably increased and more consistent when using the consensus of three annotators. Adding more annotators only resulted in minor improvements. We conclude that databases by few pathologists and high label accuracy may be the best compromise between high algorithmic performance and time investment.
Dataset on Bi- and Multi-Nucleated Tumor Cells in Canine Cutaneous Mast Cell Tumors
Jan 05, 2021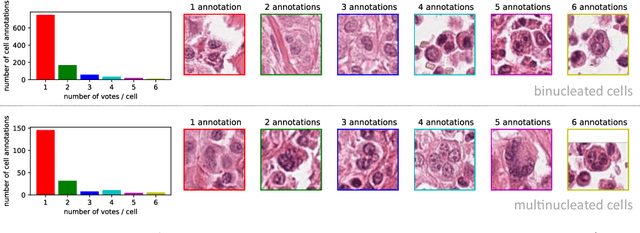
Tumor cells with two nuclei (binucleated cells, BiNC) or more nuclei (multinucleated cells, MuNC) indicate an increased amount of cellular genetic material which is thought to facilitate oncogenesis, tumor progression and treatment resistance. In canine cutaneous mast cell tumors (ccMCT), binucleation and multinucleation are parameters used in cytologic and histologic grading schemes (respectively) which correlate with poor patient outcome. For this study, we created the first open source data-set with 19,983 annotations of BiNC and 1,416 annotations of MuNC in 32 histological whole slide images of ccMCT. Labels were created by a pathologist and an algorithmic-aided labeling approach with expert review of each generated candidate. A state-of-the-art deep learning-based model yielded an $F_1$ score of 0.675 for BiNC and 0.623 for MuNC on 11 test whole slide images. In regions of interest ($2.37 mm^2$) extracted from these test images, 6 pathologists had an object detection performance between 0.270 - 0.526 for BiNC and 0.316 - 0.622 for MuNC, while our model archived an $F_1$ score of 0.667 for BiNC and 0.685 for MuNC. This open dataset can facilitate development of automated image analysis for this task and may thereby help to promote standardization of this facet of histologic tumor prognostication.
Dogs as Model for Human Breast Cancer: A Completely Annotated Whole Slide Image Dataset
Aug 24, 2020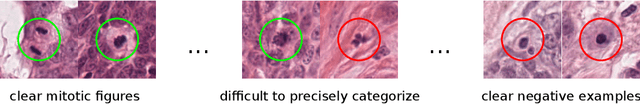
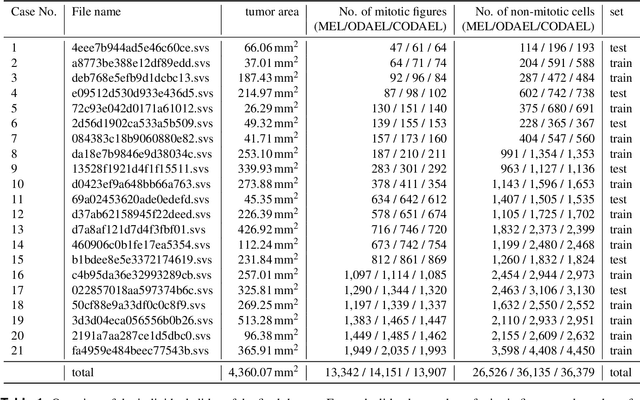
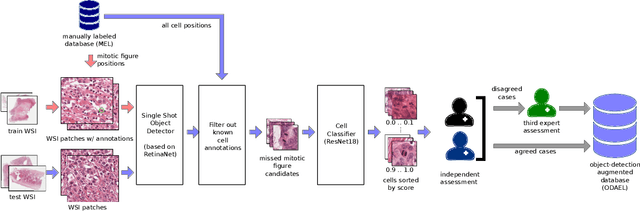
Canine mammary carcinoma (CMC) has been used as a model to investigate the pathogenesis of human breast cancer and the same grading scheme is commonly used to assess tumor malignancy in both. One key component of this grading scheme is the density of mitotic figures (MF). Current publicly available datasets on human breast cancer only provide annotations for small subsets of whole slide images (WSIs). We present a novel dataset of 21 WSIs of CMC completely annotated for MF. For this, a pathologist screened all WSIs for potential MF and structures with a similar appearance. A second expert blindly assigned labels, and for non-matching labels, a third expert assigned the final labels. Additionally, we used machine learning to identify previously undetected MF. Finally, we performed representation learning and two-dimensional projection to further increase the consistency of the annotations. Our dataset consists of 13,907 MF and 36,379 hard negatives. We achieved a mean F1-score of 0.791 on the test set and of up to 0.696 on a human breast cancer dataset.
Are pathologist-defined labels reproducible? Comparison of the TUPAC16 mitotic figure dataset with an alternative set of labels
Jul 10, 2020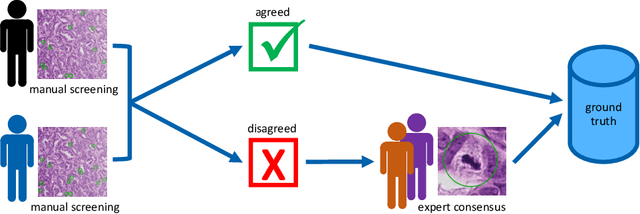
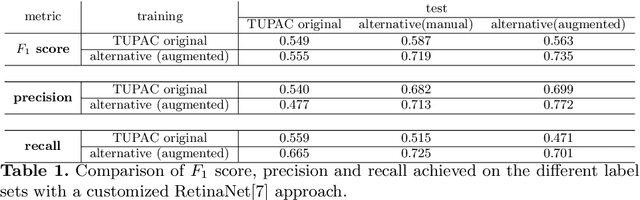
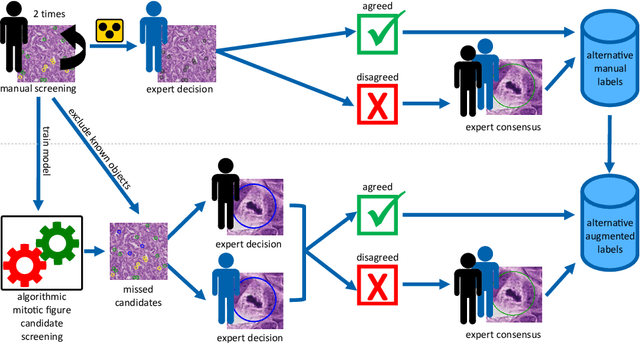
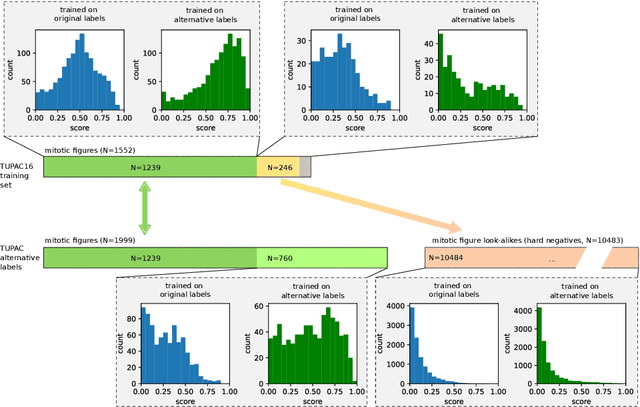
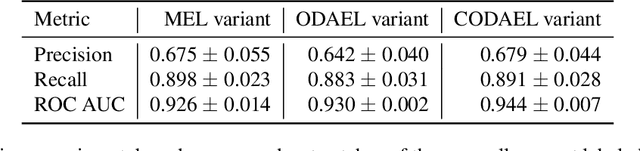
Pathologist-defined labels are the gold standard for histopathological data sets, regardless of well-known limitations in consistency for some tasks. To date, some datasets on mitotic figures are available and were used for development of promising deep learning-based algorithms. In order to assess robustness of those algorithms and reproducibility of their methods it is necessary to test on several independent datasets. The influence of different labeling methods of these available datasets is currently unknown. To tackle this, we present an alternative set of labels for the images of the auxiliary mitosis dataset of the TUPAC16 challenge. Additional to manual mitotic figure screening, we used a novel, algorithm-aided labeling process, that allowed to minimize the risk of missing rare mitotic figures in the images. All potential mitotic figures were independently assessed by two pathologists. The novel, publicly available set of labels contains 1,999 mitotic figures (+28.80%) and additionally includes 10,483 labels of cells with high similarities to mitotic figures (hard examples). We found significant difference comparing F_1 scores between the original label set (0.549) and the new alternative label set (0.735) using a standard deep learning object detection architecture. The models trained on the alternative set showed higher overall confidence values, suggesting a higher overall label consistency. Findings of the present study show that pathologists-defined labels may vary significantly resulting in notable difference in the model performance. Comparison of deep learning-based algorithms between independent datasets with different labeling methods should be done with caution.
EXACT: A collaboration toolset for algorithm-aided annotation of almost everything
Apr 30, 2020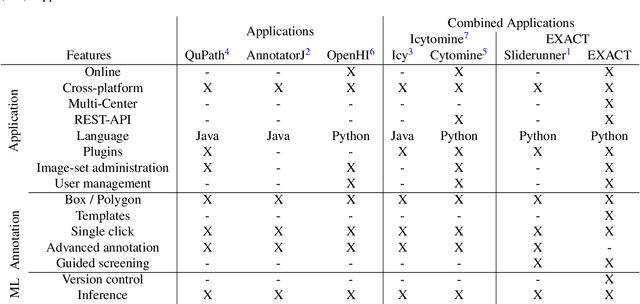
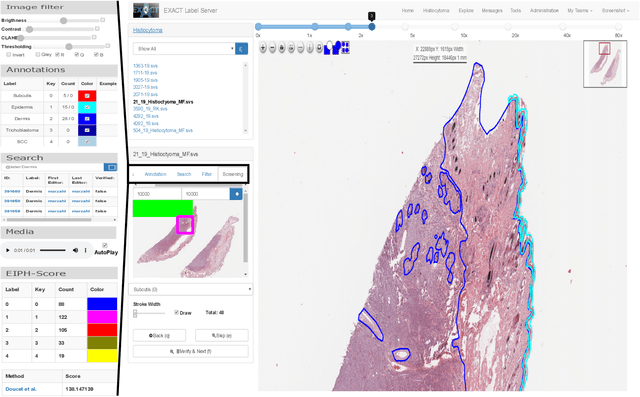
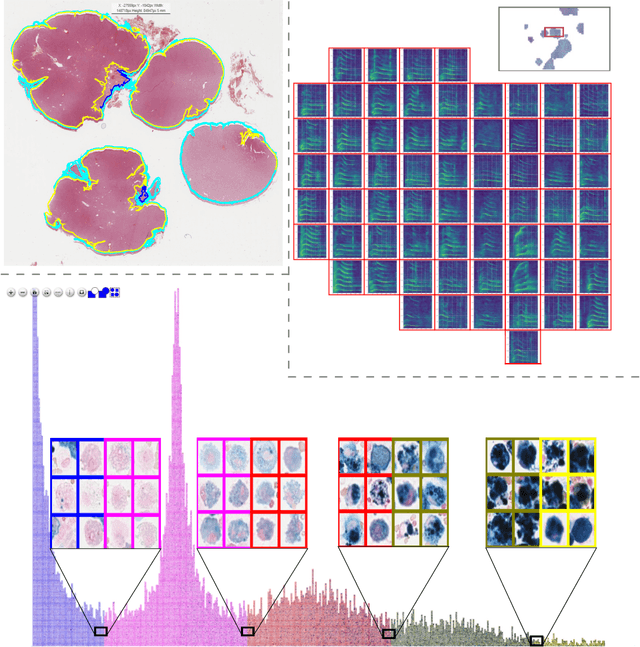
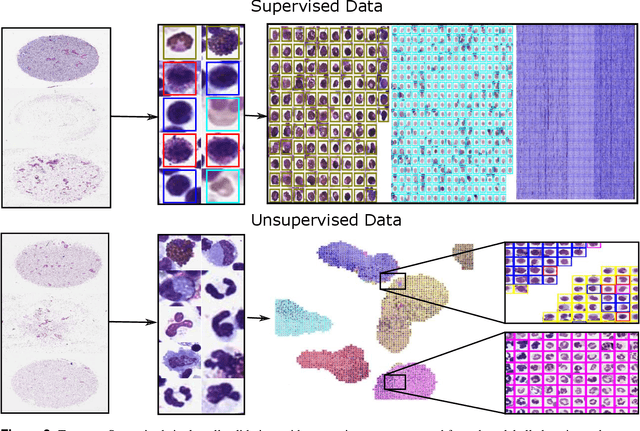
In many research areas scientific progress is accelerated by multidisciplinary access to image data and their interdisciplinary annotation. However, keeping track of these annotations to ensure a high-quality multi purpose data set is a challenging and labour intensive task. We developed the open-source online platform EXACT (EXpert Algorithm Cooperation Tool) that enables the collaborative interdisciplinary analysis of images from different domains online and offline. EXACT supports multi-gigapixel whole slide medical images, as well as image series with thousands of images. The software utilises a flexible plugin system that can be adapted to diverse applications such as counting mitotic figures with the screening mode, finding false annotations on a novel validation view, or using the latest deep learning image analysis technologies. This is combined with a version control system which makes it possible to keep track of changes in data sets and, for example, to link the results of deep learning experiments to specific data set versions. EXACT is freely available and has been applied successfully to a broad range of annotation tasks already, including highly diverse applications like deep learning supported cytology grading, interdisciplinary multi-centre whole slide image tumour annotation, and highly specialised whale sound spectroscopy clustering.
CLCNet: Deep learning-based Noise Reduction for Hearing Aids using Complex Linear Coding
Jan 28, 2020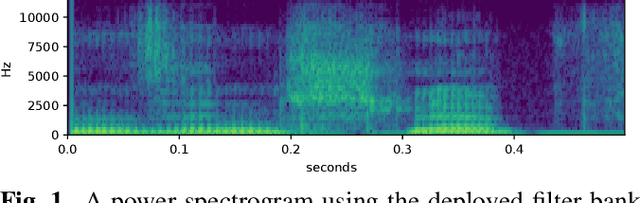
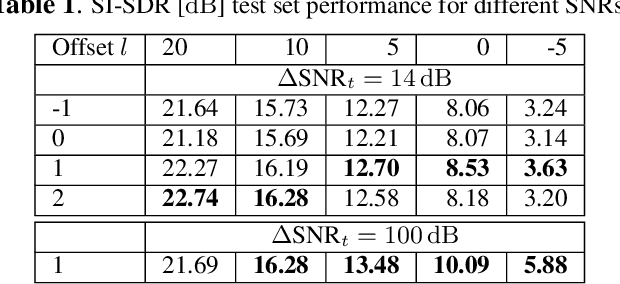
Noise reduction is an important part of modern hearing aids and is included in most commercially available devices. Deep learning-based state-of-the-art algorithms, however, either do not consider real-time and frequency resolution constrains or result in poor quality under very noisy conditions. To improve monaural speech enhancement in noisy environments, we propose CLCNet, a framework based on complex valued linear coding. First, we define complex linear coding (CLC) motivated by linear predictive coding (LPC) that is applied in the complex frequency domain. Second, we propose a framework that incorporates complex spectrogram input and coefficient output. Third, we define a parametric normalization for complex valued spectrograms that complies with low-latency and on-line processing. Our CLCNet was evaluated on a mixture of the EUROM database and a real-world noise dataset recorded with hearing aids and compared to traditional real-valued Wiener-Filter gains.
Learning New Tricks from Old Dogs -- Inter-Species, Inter-Tissue Domain Adaptation for Mitotic Figure Assessment
Nov 25, 2019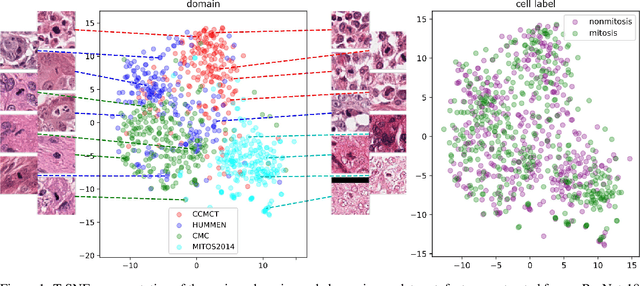
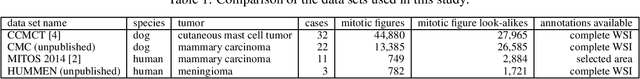
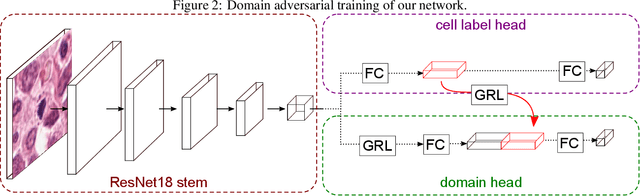
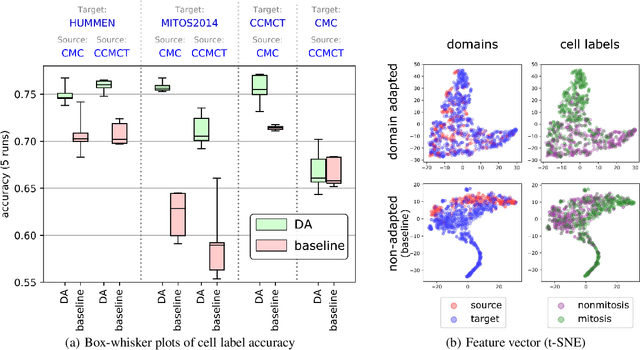
For histopathological tumor assessment, the count of mitotic figures per area is an important part of prognostication. Algorithmic approaches - such as for mitotic figure identification - have significantly improved in recent times, potentially allowing for computer-augmented or fully automatic screening systems in the future. This trend is further supported by whole slide scanning microscopes becoming available in many pathology labs and could soon become a standard imaging tool. For an application in broader fields of such algorithms, the availability of mitotic figure data sets of sufficient size for the respective tissue type and species is an important precondition, that is, however, rarely met. While algorithmic performance climbed steadily for e.g. human mammary carcinoma, thanks to several challenges held in the field, for most tumor types, data sets are not available. In this work, we assess domain transfer of mitotic figure recognition using domain adversarial training on four data sets, two from dogs and two from humans. We were able to show that domain adversarial training considerably improves accuracy when applying mitotic figure classification learned from the canine on the human data sets (up to +12.8% in accuracy) and is thus a helpful method to transfer knowledge from existing data sets to new tissue types and species.