 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvaluating Protein-protein Interaction Predictors with a Novel 3-Dimensional Metric
Nov 06, 2015
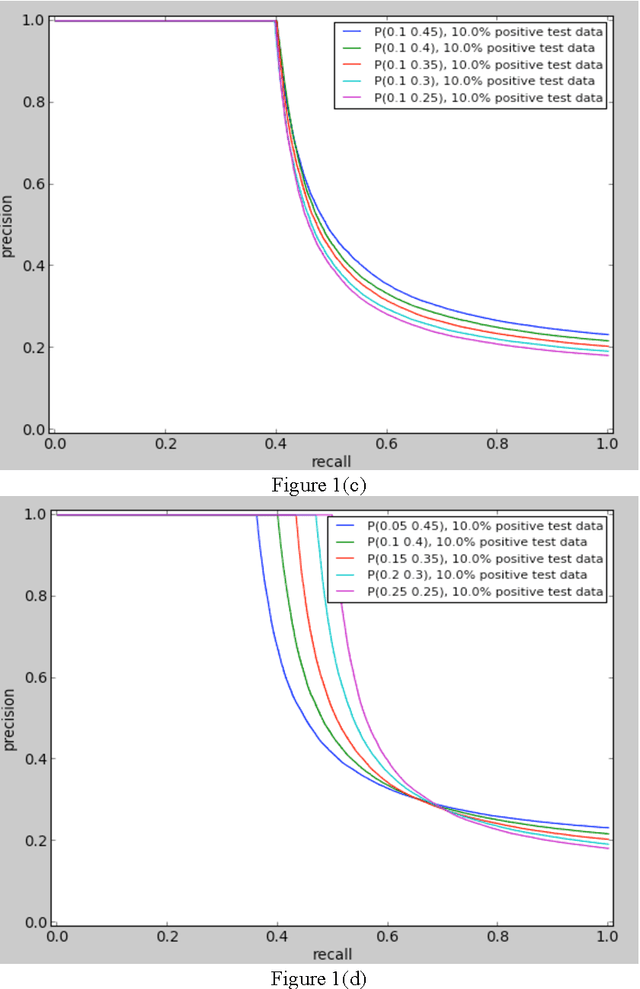
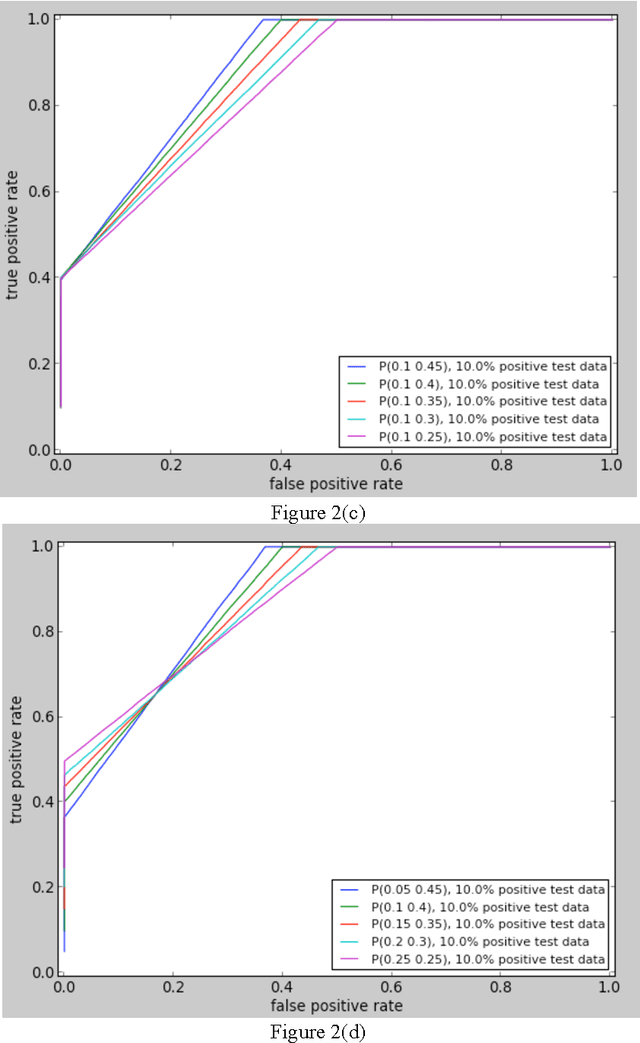
In order for the predicted interactions to be directly adopted by biologists, the ma- chine learning predictions have to be of high precision, regardless of recall. This aspect cannot be evaluated or numerically represented well by traditional metrics like accuracy, ROC, or precision-recall curve. In this work, we start from the alignment in sensitivity of ROC and recall of precision-recall curve, and propose an evaluation metric focusing on the ability of a model to be adopted by biologists. This metric evaluates the ability of a machine learning algorithm to predict only new interactions, meanwhile, it eliminates the influence of test dataset. In the experiment of evaluating different classifiers with a same data set and evaluating the same predictor with different datasets, our new metric fulfills the evaluation task of our interest while two widely recognized metrics, ROC and precision-recall curve fail the tasks for different reasons.
Evaluation of Protein-protein Interaction Predictors with Noisy Partially Labeled Data Sets
Sep 18, 2015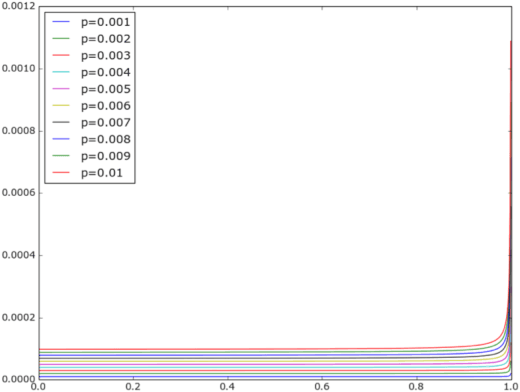

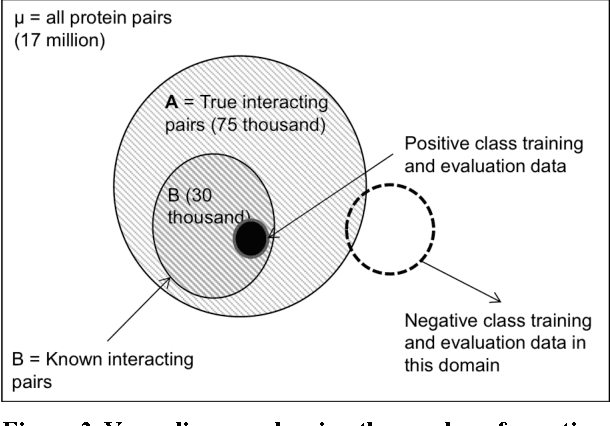

Protein-protein interaction (PPI) prediction is an important problem in machine learning and computational biology. However, there is no data set for training or evaluation purposes, where all the instances are accurately labeled. Instead, what is available are instances of positive class (with possibly noisy labels) and no instances of negative class. The non-availability of negative class data is typically handled with the observation that randomly chosen protein-pairs have a nearly 100% chance of being negative class, as only 1 in 1,500 protein pairs expected is expected to be an interacting pair. In this paper, we focused on the problem that non-availability of accurately labeled testing data sets in the domain of protein-protein interaction (PPI) prediction may lead to biased evaluation results. We first showed that not acknowledging the inherent skew in the interactome (i.e. rare occurrence of positive instances) leads to an over-estimated accuracy of the predictor. Then we show that, with the belief that positive interactions are a rare category, sampling random pairs of proteins excluding known interacting proteins set as the negative testing data set could lead to an under-estimated evaluation result. We formalized those two problems to validate the above claim, and based on the formalization, we proposed a balancing method to cancel out the over-estimation with under-estimation. Finally, our experiments validated the theoretical aspects and showed that this balancing evaluation could evaluate the exact performance without availability of golden standard data sets.