 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBreaking the bonds of generative artificial intelligence by minimizing the maximum entropy
Feb 18, 2025The emergence of generative artificial intelligence (GenAI), comprising large language models, text-to-image generators, and AI algorithms for medical drug and material design, had a transformative impact on society. However, despite an initial exponential growth surpassing Moore's law, progress is now plateauing, suggesting we are approaching the limits of current technology. Indeed, these models are notoriously data-hungry, prone to overfitting, and challenging to direct during the generative process, hampering their effective professional employment. To cope with these limitations, we propose a paradigm shift in GenAI by introducing an ab initio method based on the minimal maximum entropy principle. Our approach does not fit the data. Instead, it compresses information in the training set by finding a latent representation parameterized by arbitrary nonlinear functions, such as neural networks. The result is a general physics-driven model, which is data-efficient, resistant to overfitting, and flexible, permitting to control and influence the generative process. Benchmarking shows that our method outperforms variational autoencoders (VAEs) with similar neural architectures, particularly on undersampled datasets. We demonstrate the methods effectiveness in generating images, even with limited training data, and its unprecedented capability to customize the generation process a posteriori without the need of any fine-tuning or retraining.
Understanding Anharmonic Effects on Hydrogen Desorption Characteristics of Mg$_n$H$_{2n}$ Nanoclusters by ab initio trained Deep Neural Network
Nov 27, 2021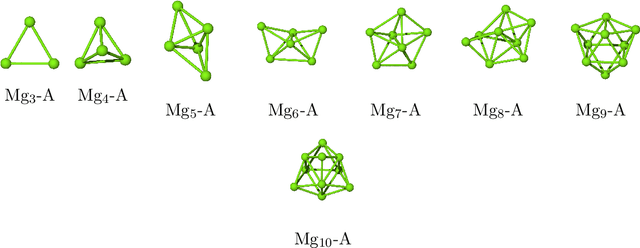
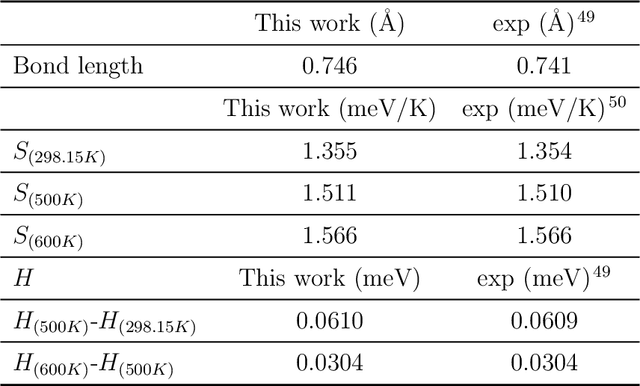
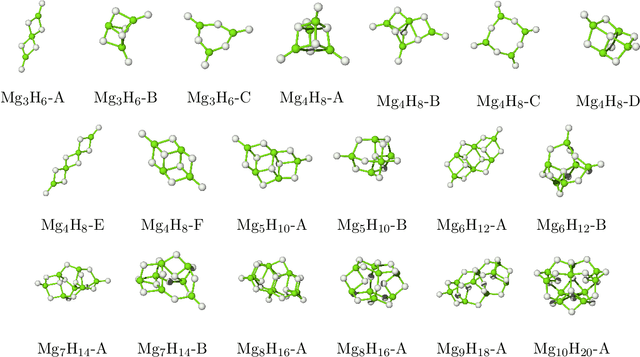
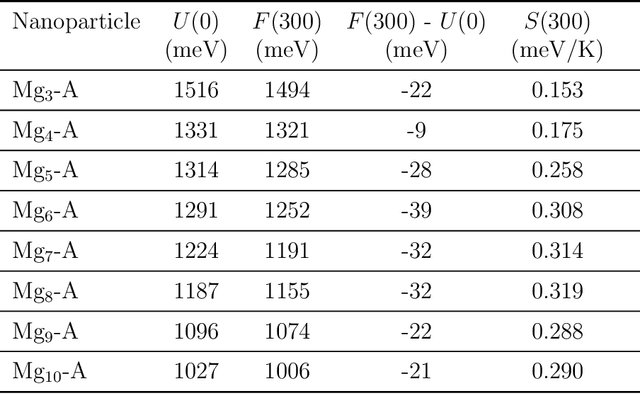
Magnesium hydride (MgH$_2$) has been widely studied for effective hydrogen storage. However, its bulk desorption temperature (553 K) is deemed too high for practical applications. Besides doping, a strategy to decrease such reaction energy for releasing hydrogen is the use of MgH$_2$-based nanoparticles (NPs). Here, we investigate first the thermodynamic properties of Mg$_n$H$_{2n}$ NPs ($n<10$) from first-principles, in particular by assessing the anharmonic effects on the enthalpy, entropy and thermal expansion by means of the Stochastic Self Consistent Harmonic Approximation (SSCHA). The latter method goes beyond previous approaches, typically based on molecular mechanics and the quasi-harmonic approximation, allowing the ab initio calculation of the fully-anharmonic free energy. We find an almost linear dependence on temperature of the interatomic bond lengths - with a relative variation of few percent over 300K -, alongside with a bond distance decrease of the Mg-H bonds. In order to increase the size of NPs toward experiments of hydrogen desorption from MgH$_2$ we devise a computationally effective Machine Learning model trained to accurately determine the forces and total energies (i.e. the potential energy surfaces), integrating the latter with the SSCHA model to fully include the anharmonic effects. We find a significative decrease of the H-desorption temperature for sub-nanometric clusters Mg$_n$H$_{2n}$ with $n \leq 10$, with a non-negligible, although little effect due to anharmonicities (up to 10%).