 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEmulating Radiative Transfer in Astrophysical Environments
Nov 11, 2025Radiative transfer is a fundamental process in astrophysics, essential for both interpreting observations and modeling thermal and dynamical feedback in simulations via ionizing radiation and photon pressure. However, numerically solving the underlying radiative transfer equation is computationally intensive due to the complex interaction of light with matter and the disparity between the speed of light and the typical gas velocities in astrophysical environments, making it particularly expensive to include the effects of on-the-fly radiation in hydrodynamic simulations. This motivates the development of surrogate models that can significantly accelerate radiative transfer calculations while preserving high accuracy. We present a surrogate model based on a Fourier Neural Operator architecture combined with U-Nets. Our model approximates three-dimensional, monochromatic radiative transfer in time-dependent regimes, in absorption-emission approximation, achieving speedups of more than 2 orders of magnitude while maintaining an average relative error below 3%, demonstrating our approach's potential to be integrated into state-of-the-art hydrodynamic simulations.
Emulating the interstellar medium chemistry with neural operators
Feb 19, 2024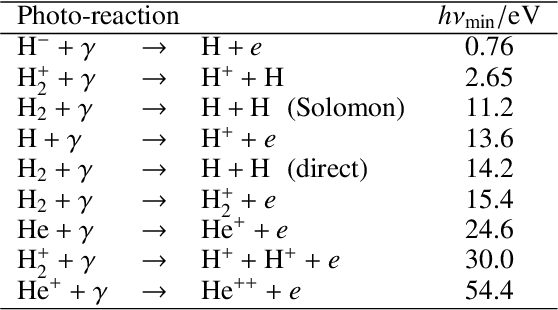
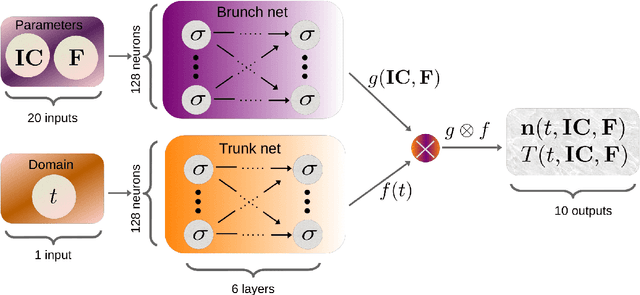

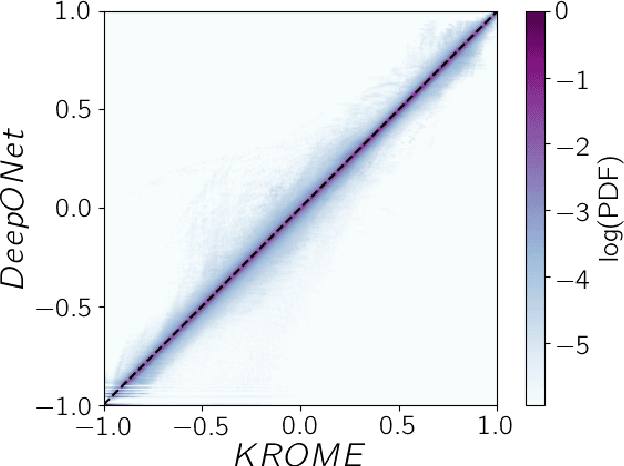
Galaxy formation and evolution critically depend on understanding the complex photo-chemical processes that govern the evolution and thermodynamics of the InterStellar Medium (ISM). Computationally, solving chemistry is among the most heavy tasks in cosmological and astrophysical simulations. The evolution of such non-equilibrium photo-chemical network relies on implicit, precise, computationally costly, ordinary differential equations (ODE) solvers. Here, we aim at substituting such procedural solvers with fast, pre-trained, emulators based on neural operators. We emulate a non-equilibrium chemical network up to H$_2$ formation (9 species, 52 reactions) by adopting the DeepONet formalism, i.e. by splitting the ODE solver operator that maps the initial conditions and time evolution into a tensor product of two neural networks. We use $\texttt{KROME}$ to generate a training set spanning $-2\leq \log(n/\mathrm{cm}^{-3}) \leq 3.5$, $\log(20) \leq\log(T/\mathrm{K}) \leq 5.5$, $-6 \leq \log(n_i/n) < 0$, and by adopting an incident radiation field $\textbf{F}$ sampled in 10 energy bins with a continuity prior. We separately train the solver for $T$ and each $n_i$ for $\simeq 4.34\,\rm GPUhrs$. Compared with the reference solutions obtained by $\texttt{KROME}$ for single zone models, the typical precision obtained is of order $10^{-2}$, i.e. the $10 \times$ better with a training that is $40 \times$ less costly with respect to previous emulators which however considered only a fixed $\mathbf{F}$. The present model achieves a speed-up of a factor of $128 \times$ with respect to stiff ODE solvers. Our neural emulator represents a significant leap forward in the modeling of ISM chemistry, offering a good balance of precision, versatility, and computational efficiency.
Neural networks: solving the chemistry of the interstellar medium
Nov 28, 2022Non-equilibrium chemistry is a key process in the study of the InterStellar Medium (ISM), in particular the formation of molecular clouds and thus stars. However, computationally it is among the most difficult tasks to include in astrophysical simulations, because of the typically high (>40) number of reactions, the short evolutionary timescales (about $10^4$ times less than the ISM dynamical time) and the characteristic non-linearity and stiffness of the associated Ordinary Differential Equations system (ODEs). In this proof of concept work, we show that Physics Informed Neural Networks (PINN) are a viable alternative to traditional ODE time integrators for stiff thermo-chemical systems, i.e. up to molecular hydrogen formation (9 species and 46 reactions). Testing different chemical networks in a wide range of densities ($-2< \log n/{\rm cm}^{-3}< 3$) and temperatures ($1 < \log T/{\rm K}< 5$), we find that a basic architecture can give a comfortable convergence only for simplified chemical systems: to properly capture the sudden chemical and thermal variations a Deep Galerkin Method is needed. Once trained ($\sim 10^3$ GPUhr), the PINN well reproduces the strong non-linear nature of the solutions (errors $\lesssim 10\%$) and can give speed-ups up to a factor of $\sim 200$ with respect to traditional ODE solvers. Further, the latter have completion times that vary by about $\sim 30\%$ for different initial $n$ and $T$, while the PINN method gives negligible variations. Both the speed-up and the potential improvement in load balancing imply that PINN-powered simulations are a very palatable way to solve complex chemical calculation in astrophysical and cosmological problems.