 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhancing DNA Foundation Models to Address Masking Inefficiencies
Feb 25, 2025Masked language modelling (MLM) as a pretraining objective has been widely adopted in genomic sequence modelling. While pretrained models can successfully serve as encoders for various downstream tasks, the distribution shift between pretraining and inference detrimentally impacts performance, as the pretraining task is to map [MASK] tokens to predictions, yet the [MASK] is absent during downstream applications. This means the encoder does not prioritize its encodings of non-[MASK] tokens, and expends parameters and compute on work only relevant to the MLM task, despite this being irrelevant at deployment time. In this work, we propose a modified encoder-decoder architecture based on the masked autoencoder framework, designed to address this inefficiency within a BERT-based transformer. We empirically show that the resulting mismatch is particularly detrimental in genomic pipelines where models are often used for feature extraction without fine-tuning. We evaluate our approach on the BIOSCAN-5M dataset, comprising over 2 million unique DNA barcodes. We achieve substantial performance gains in both closed-world and open-world classification tasks when compared against causal models and bidirectional architectures pretrained with MLM tasks.
CGRclust: Chaos Game Representation for Twin Contrastive Clustering of Unlabelled DNA Sequences
Jul 01, 2024This study proposes CGRclust, a novel combination of unsupervised twin contrastive clustering of Chaos Game Representations (CGR) of DNA sequences, with convolutional neural networks (CNNs). To the best of our knowledge, CGRclust is the first method to use unsupervised learning for image classification (herein applied to two-dimensional CGR images) for clustering datasets of DNA sequences. CGRclust overcomes the limitations of traditional sequence classification methods by leveraging unsupervised twin contrastive learning to detect distinctive sequence patterns, without requiring DNA sequence alignment or biological/taxonomic labels. CGRclust accurately clustered twenty-five diverse datasets, with sequence lengths ranging from 664 bp to 100 kbp, including mitochondrial genomes of fish, fungi, and protists, as well as viral whole genome assemblies and synthetic DNA sequences. Compared with three recent clustering methods for DNA sequences (DeLUCS, iDeLUCS, and MeShClust v3.0.), CGRclust is the only method that surpasses 81.70% accuracy across all four taxonomic levels tested for mitochondrial DNA genomes of fish. Moreover, CGRclust also consistently demonstrates superior performance across all the viral genomic datasets. The high clustering accuracy of CGRclust on these twenty-five datasets, which vary significantly in terms of sequence length, number of genomes, number of clusters, and level of taxonomy, demonstrates its robustness, scalability, and versatility.
BIOSCAN-5M: A Multimodal Dataset for Insect Biodiversity
Jun 18, 2024



As part of an ongoing worldwide effort to comprehend and monitor insect biodiversity, this paper presents the BIOSCAN-5M Insect dataset to the machine learning community and establish several benchmark tasks. BIOSCAN-5M is a comprehensive dataset containing multi-modal information for over 5 million insect specimens, and it significantly expands existing image-based biological datasets by including taxonomic labels, raw nucleotide barcode sequences, assigned barcode index numbers, and geographical information. We propose three benchmark experiments to demonstrate the impact of the multi-modal data types on the classification and clustering accuracy. First, we pretrain a masked language model on the DNA barcode sequences of the \mbox{BIOSCAN-5M} dataset, and demonstrate the impact of using this large reference library on species- and genus-level classification performance. Second, we propose a zero-shot transfer learning task applied to images and DNA barcodes to cluster feature embeddings obtained from self-supervised learning, to investigate whether meaningful clusters can be derived from these representation embeddings. Third, we benchmark multi-modality by performing contrastive learning on DNA barcodes, image data, and taxonomic information. This yields a general shared embedding space enabling taxonomic classification using multiple types of information and modalities. The code repository of the BIOSCAN-5M Insect dataset is available at {\url{https://github.com/zahrag/BIOSCAN-5M}}
BarcodeBERT: Transformers for Biodiversity Analysis
Nov 04, 2023



Understanding biodiversity is a global challenge, in which DNA barcodes - short snippets of DNA that cluster by species - play a pivotal role. In particular, invertebrates, a highly diverse and under-explored group, pose unique taxonomic complexities. We explore machine learning approaches, comparing supervised CNNs, fine-tuned foundation models, and a DNA barcode-specific masking strategy across datasets of varying complexity. While simpler datasets and tasks favor supervised CNNs or fine-tuned transformers, challenging species-level identification demands a paradigm shift towards self-supervised pretraining. We propose BarcodeBERT, the first self-supervised method for general biodiversity analysis, leveraging a 1.5 M invertebrate DNA barcode reference library. This work highlights how dataset specifics and coverage impact model selection, and underscores the role of self-supervised pretraining in achieving high-accuracy DNA barcode-based identification at the species and genus level. Indeed, without the fine-tuning step, BarcodeBERT pretrained on a large DNA barcode dataset outperforms DNABERT and DNABERT-2 on multiple downstream classification tasks. The code repository is available at https://github.com/Kari-Genomics-Lab/BarcodeBERT
Map of Life: Measuring and Visualizing Species' Relatedness with "Molecular Distance Maps"
Jul 14, 2013
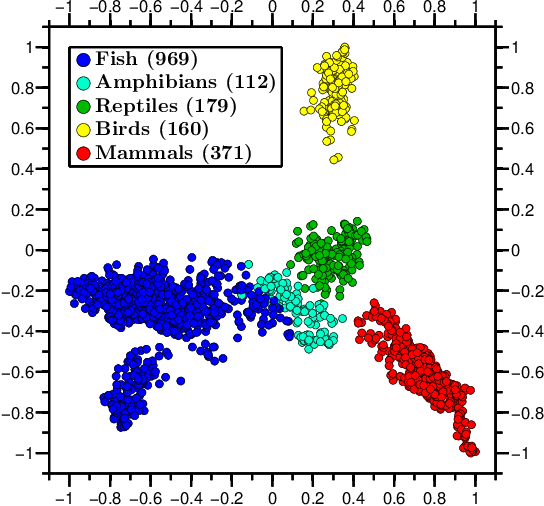
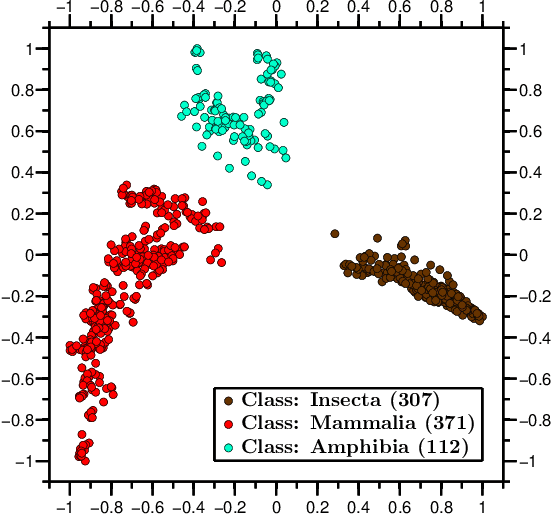
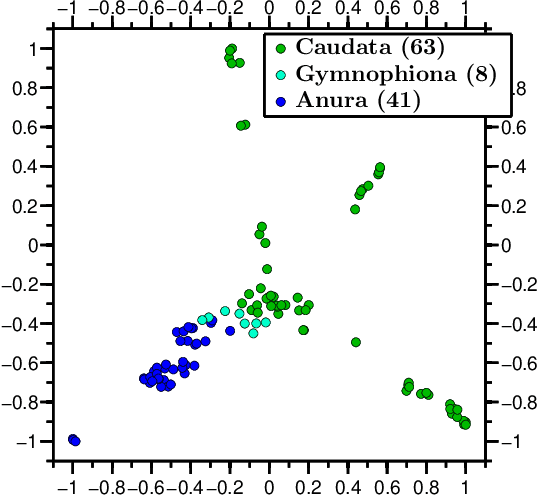
We propose a novel combination of methods that (i) portrays quantitative characteristics of a DNA sequence as an image, (ii) computes distances between these images, and (iii) uses these distances to output a map wherein each sequence is a point in a common Euclidean space. In the resulting "Molecular Distance Map" each point signifies a DNA sequence, and the geometric distance between any two points reflects the degree of relatedness between the corresponding sequences and species. Molecular Distance Maps present compelling visual representations of relationships between species and could be used for taxonomic clarifications, for species identification, and for studies of evolutionary history. One of the advantages of this method is its general applicability since, as sequence alignment is not required, the DNA sequences chosen for comparison can be completely different regions in different genomes. In fact, this method can be used to compare any two DNA sequences. For example, in our dataset of 3,176 mitochondrial DNA sequences, it correctly finds the mtDNA sequences most closely related to that of the anatomically modern human (the Neanderthal, the Denisovan, and the chimp), and it finds that the sequence most different from it belongs to a cucumber. Furthermore, our method can be used to compare real sequences to artificial, computer-generated, DNA sequences. For example, it is used to determine that the distances between a Homo sapiens sapiens mtDNA and artificial sequences of the same length and same trinucleotide frequencies can be larger than the distance between the same human mtDNA and the mtDNA of a fruit-fly. We demonstrate this method's promising potential for taxonomical clarifications by applying it to a diverse variety of cases that have been historically controversial, such as the genus Polypterus, the family Tarsiidae, and the vast (super)kingdom Protista.