 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAmorProt: Amino Acid Molecular Fingerprints Repurposing based Protein Fingerprint
Mar 27, 2023As protein therapeutics play an important role in almost all medical fields, numerous studies have been conducted on proteins using artificial intelligence. Artificial intelligence has enabled data driven predictions without the need for expensive experiments. Nevertheless, unlike the various molecular fingerprint algorithms that have been developed, protein fingerprint algorithms have rarely been studied. In this study, we proposed the amino acid molecular fingerprints repurposing based protein (AmorProt) fingerprint, a protein sequence representation method that effectively uses the molecular fingerprints corresponding to 20 amino acids. Subsequently, the performances of the tree based machine learning and artificial neural network models were compared using (1) amyloid classification and (2) isoelectric point regression. Finally, the applicability and advantages of the developed platform were demonstrated through a case study and the following experiments: (3) comparison of dataset dependence with feature based methods; (4) feature importance analysis; and (5) protein space analysis. Consequently, the significantly improved model performance and data set independent versatility of the AmorProt fingerprint were verified. The results revealed that the current protein representation method can be applied to various fields related to proteins, such as predicting their fundamental properties or interaction with ligands.
Machine Learning-Aided Discovery of Superionic Solid-State Electrolyte for Li-Ion Batteries
Feb 14, 2022



Li-Ion Solid-State Electrolytes (Li-SSEs) are a promising solution that resolves the critical issues of conventional Li-Ion Batteries (LIBs) such as poor ionic conductivity, interfacial instability, and dendrites growth. In this study, a platform consisting of a high-throughput screening and a machine-learning surrogate model for discovering superionic Li-SSEs among 20,237 Li-containing materials is developed. For the training database, the ionic conductivity of Na SuperIonic CONductor (NASICON) and Li SuperIonic CONductor (LISICON) type SSEs are obtained from the previous literature. Then, the chemical descriptor (CD) and additional structural properties are used as machine-readable features. Li-SSE candidates are selected through the screening criteria, and the prediction on the ionic conductivity of those is followed. Then, to reduce uncertainty in the surrogate model, the ensemble method by considering the best-performing two models is employed, whose mean prediction accuracy is 0.843 and 0.829, respectively. Furthermore, first-principles calculations are conducted for confirming the ionic conductivity of the strong candidates. Finally, six potential superionic Li-SSEs that have not previously been investigated are proposed. We believe that the constructed platform can accelerate the search for Li-SSEs with high ionic conductivity at minimum cost.
MGCVAE: Multi-objective Inverse Design via Molecular Graph Conditional Variational Autoencoder
Feb 14, 2022
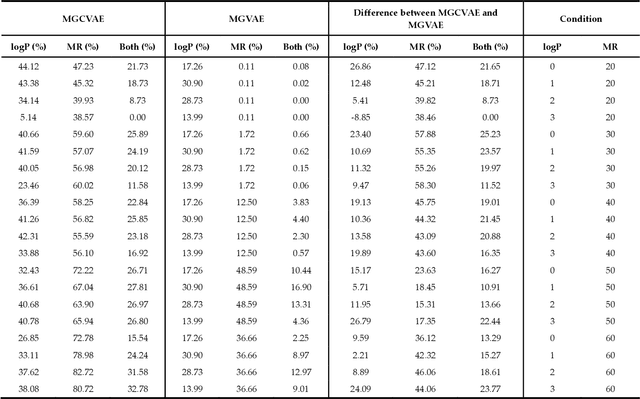
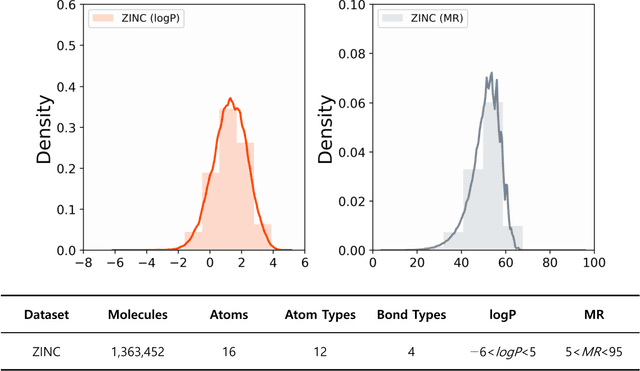

The ultimate goal of various fields is to directly generate molecules with desired properties, such as finding water-soluble molecules in drug development and finding molecules suitable for organic light-emitting diode (OLED) or photosensitizers in the field of development of new organic materials. In this respect, this study proposes a molecular graph generative model based on the autoencoder for de novo design. The performance of molecular graph conditional variational autoencoder (MGCVAE) for generating molecules having specific desired properties is investigated by comparing it to molecular graph variational autoencoder (MGVAE). Furthermore, multi-objective optimization for MGCVAE was applied to satisfy two selected properties simultaneously. In this study, two physical properties -- logP and molar refractivity -- were used as optimization targets for the purpose of designing de novo molecules, especially in drug discovery. As a result, it was confirmed that among generated molecules, 25.89% optimized molecules were generated in MGCVAE compared to 0.66% in MGVAE. Hence, it demonstrates that MGCVAE effectively produced drug-like molecules with two target properties. The results of this study suggest that these graph-based data-driven models are one of the effective methods of designing new molecules that fulfill various physical properties, such as drug discovery.