 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePaper2Agent: Reimagining Research Papers As Interactive and Reliable AI Agents
Sep 08, 2025We introduce Paper2Agent, an automated framework that converts research papers into AI agents. Paper2Agent transforms research output from passive artifacts into active systems that can accelerate downstream use, adoption, and discovery. Conventional research papers require readers to invest substantial effort to understand and adapt a paper's code, data, and methods to their own work, creating barriers to dissemination and reuse. Paper2Agent addresses this challenge by automatically converting a paper into an AI agent that acts as a knowledgeable research assistant. It systematically analyzes the paper and the associated codebase using multiple agents to construct a Model Context Protocol (MCP) server, then iteratively generates and runs tests to refine and robustify the resulting MCP. These paper MCPs can then be flexibly connected to a chat agent (e.g. Claude Code) to carry out complex scientific queries through natural language while invoking tools and workflows from the original paper. We demonstrate Paper2Agent's effectiveness in creating reliable and capable paper agents through in-depth case studies. Paper2Agent created an agent that leverages AlphaGenome to interpret genomic variants and agents based on ScanPy and TISSUE to carry out single-cell and spatial transcriptomics analyses. We validate that these paper agents can reproduce the original paper's results and can correctly carry out novel user queries. By turning static papers into dynamic, interactive AI agents, Paper2Agent introduces a new paradigm for knowledge dissemination and a foundation for the collaborative ecosystem of AI co-scientists.
Learning Causal Representations of Single Cells via Sparse Mechanism Shift Modeling
Nov 09, 2022



Latent variable models such as the Variational Auto-Encoder (VAE) have become a go-to tool for analyzing biological data, especially in the field of single-cell genomics. One remaining challenge is the interpretability of latent variables as biological processes that define a cell's identity. Outside of biological applications, this problem is commonly referred to as learning disentangled representations. Although several disentanglement-promoting variants of the VAE were introduced, and applied to single-cell genomics data, this task has been shown to be infeasible from independent and identically distributed measurements, without additional structure. Instead, recent methods propose to leverage non-stationary data, as well as the sparse mechanism shift assumption in order to learn disentangled representations with a causal semantic. Here, we extend the application of these methodological advances to the analysis of single-cell genomics data with genetic or chemical perturbations. More precisely, we propose a deep generative model of single-cell gene expression data for which each perturbation is treated as a stochastic intervention targeting an unknown, but sparse, subset of latent variables. We benchmark these methods on simulated single-cell data to evaluate their performance at latent units recovery, causal target identification and out-of-domain generalization. Finally, we apply those approaches to two real-world large-scale gene perturbation data sets and find that models that exploit the sparse mechanism shift hypothesis surpass contemporary methods on a transfer learning task. We implement our new model and benchmarks using the scvi-tools library, and release it as open-source software at https://github.com/Genentech/sVAE.
Large-Scale Differentiable Causal Discovery of Factor Graphs
Jun 15, 2022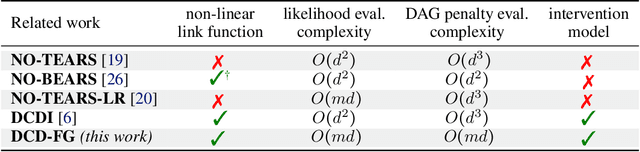
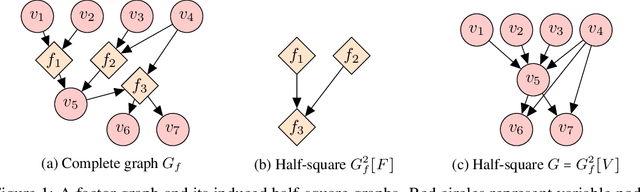
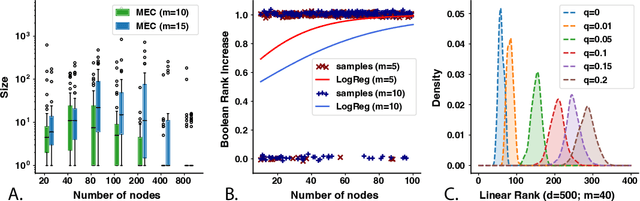

A common theme in causal inference is learning causal relationships between observed variables, also known as causal discovery. This is usually a daunting task, given the large number of candidate causal graphs and the combinatorial nature of the search space. Perhaps for this reason, most research has so far focused on relatively small causal graphs, with up to hundreds of nodes. However, recent advances in fields like biology enable generating experimental data sets with thousands of interventions followed by rich profiling of thousands of variables, raising the opportunity and urgent need for large causal graph models. Here, we introduce the notion of factor directed acyclic graphs (f-DAGs) as a way to restrict the search space to non-linear low-rank causal interaction models. Combining this novel structural assumption with recent advances that bridge the gap between causal discovery and continuous optimization, we achieve causal discovery on thousands of variables. Additionally, as a model for the impact of statistical noise on this estimation procedure, we study a model of edge perturbations of the f-DAG skeleton based on random graphs and quantify the effect of such perturbations on the f-DAG rank. This theoretical analysis suggests that the set of candidate f-DAGs is much smaller than the whole DAG space and thus more statistically robust in the high-dimensional regime where the underlying skeleton is hard to assess. We propose Differentiable Causal Discovery of Factor Graphs (DCD-FG), a scalable implementation of f-DAG constrained causal discovery for high-dimensional interventional data. DCD-FG uses a Gaussian non-linear low-rank structural equation model and shows significant improvements compared to state-of-the-art methods in both simulations as well as a recent large-scale single-cell RNA sequencing data set with hundreds of genetic interventions.