 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIdentifying Structures in Social Conversations in NSCLC Patients through the Semi-Automatic extraction of Topical Taxonomies
Feb 12, 2016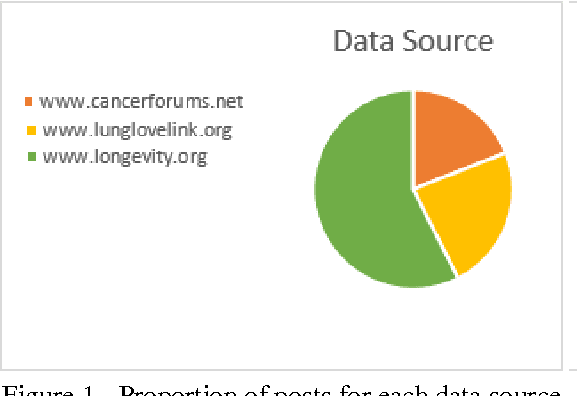
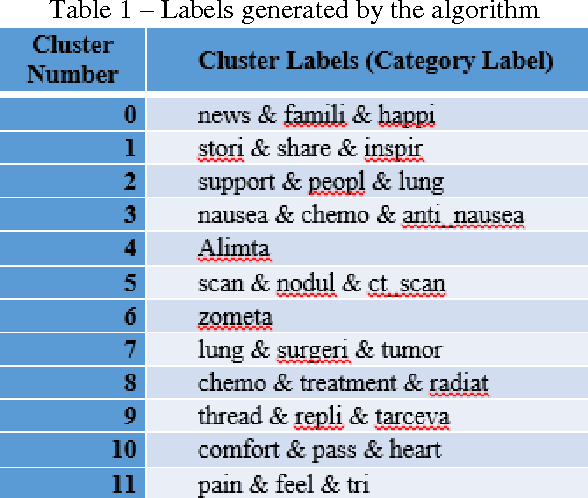
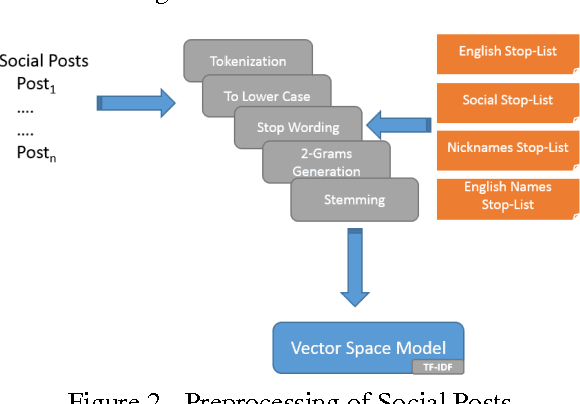
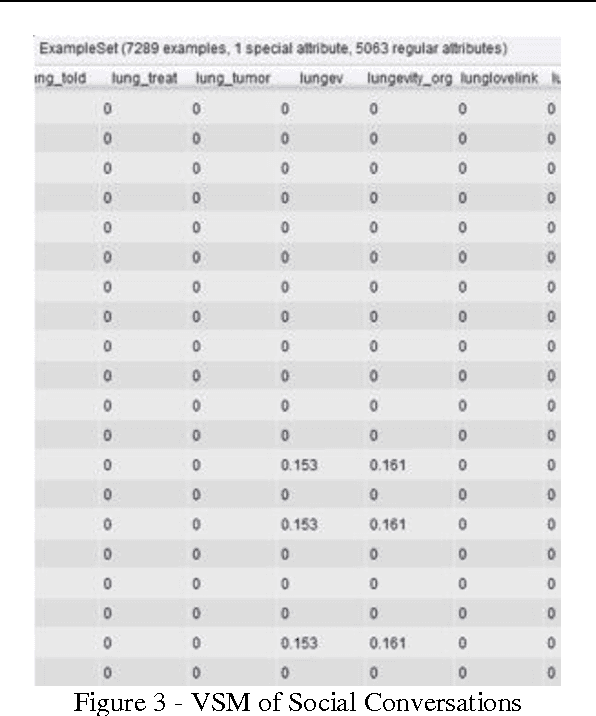
The exploration of social conversations for addressing patient's needs is an important analytical task in which many scholarly publications are contributing to fill the knowledge gap in this area. The main difficulty remains the inability to turn such contributions into pragmatic processes the pharmaceutical industry can leverage in order to generate insight from social media data, which can be considered as one of the most challenging source of information available today due to its sheer volume and noise. This study is based on the work by Scott Spangler and Jeffrey Kreulen and applies it to identify structure in social media through the extraction of a topical taxonomy able to capture the latent knowledge in social conversations in health-related sites. The mechanism for automatically identifying and generating a taxonomy from social conversations is developed and pressured tested using public data from media sites focused on the needs of cancer patients and their families. Moreover, a novel method for generating the category's label and the determination of an optimal number of categories is presented which extends Scott and Jeffrey's research in a meaningful way. We assume the reader is familiar with taxonomies, what they are and how they are used.
* 7 pages, 7 figures, 1 table
Using Ensemble Models in the Histological Examination of Tissue Abnormalities
May 15, 2015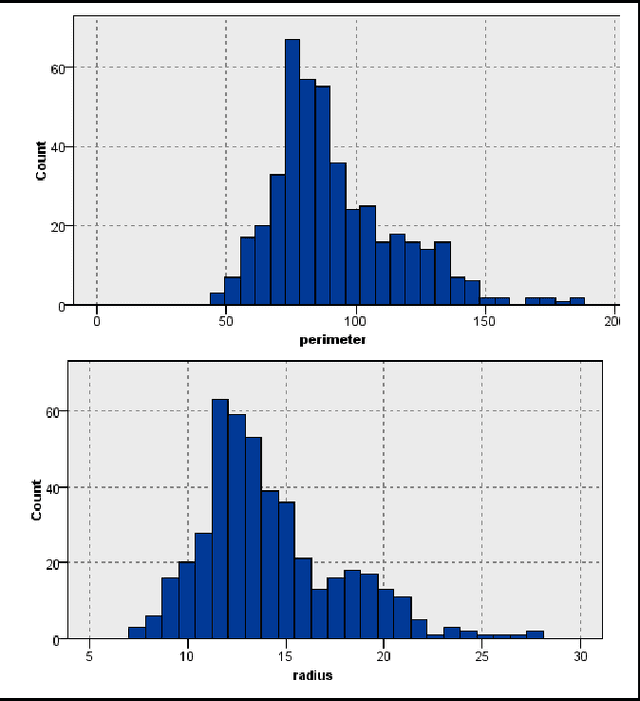
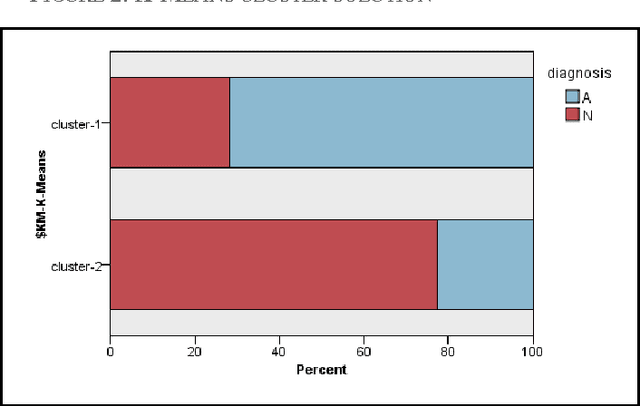
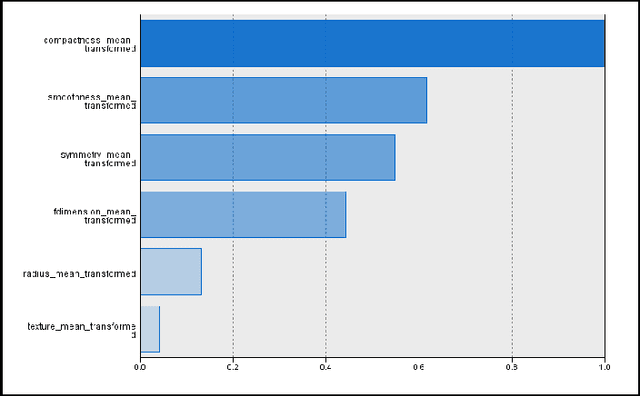
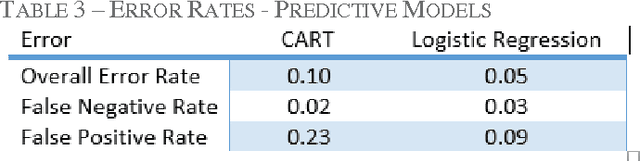
Classification models for the automatic detection of abnormalities on histological samples do exists, with an active debate on the cost associated with false negative diagnosis (underdiagnosis) and false positive diagnosis (overdiagnosis). Current models tend to underdiagnose, failing to recognize a potentially fatal disease. The objective of this study is to investigate the possibility of automatically identifying abnormalities in tissue samples through the use of an ensemble model on data generated by histological examination and to minimize the number of false negative cases.
A Multivariate Biomarker for Parkinson's Disease
May 15, 2015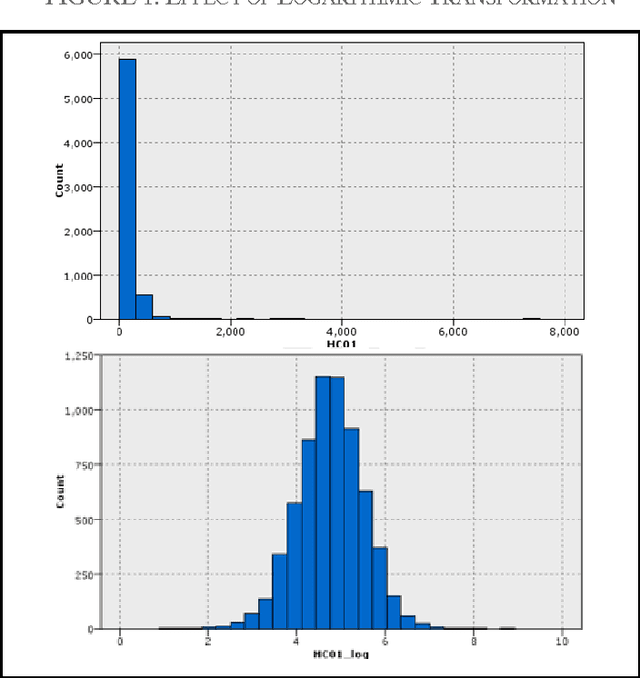
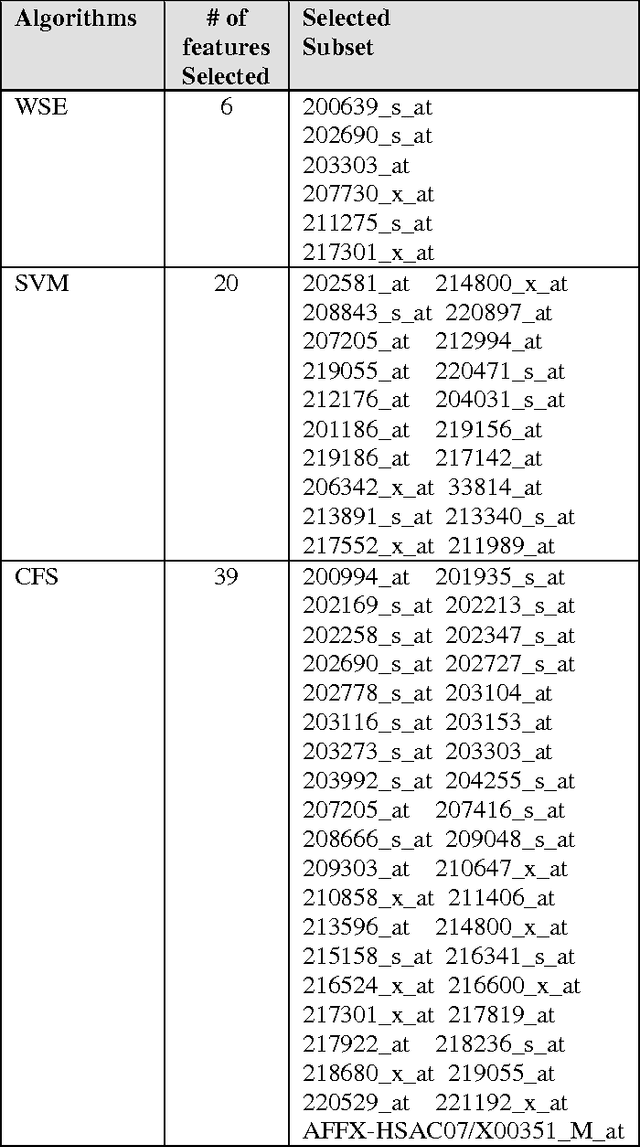
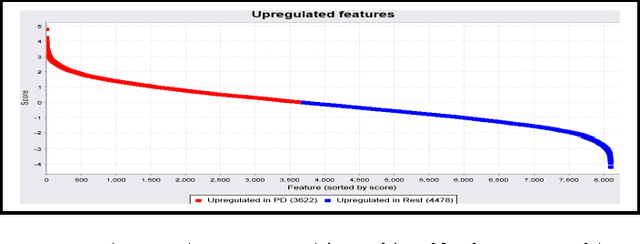

In this study, we executed a genomic analysis with the objective of selecting a set of genes (possibly small) that would help in the detection and classification of samples from patients affected by Parkinson Disease. We performed a complete data analysis and during the exploratory phase, we selected a list of differentially expressed genes. Despite their association with the diseased state, we could not use them as a biomarker tool. Therefore, our research was extended to include a multivariate analysis approach resulting in the identification and selection of a group of 20 genes that showed a clear potential in detecting and correctly classify Parkinson Disease samples even in the presence of other neurodegenerative disorders.