 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLoop-Diffusion: an equivariant diffusion model for designing and scoring protein loops
Sep 26, 2024

Predicting protein functional characteristics from structure remains a central problem in protein science, with broad implications from understanding the mechanisms of disease to designing novel therapeutics. Unfortunately, current machine learning methods are limited by scarce and biased experimental data, and physics-based methods are either too slow to be useful, or too simplified to be accurate. In this work, we present Loop-Diffusion, an energy based diffusion model which leverages a dataset of general protein loops from the entire protein universe to learn an energy function that generalizes to functional prediction tasks. We evaluate Loop-Diffusion's performance on scoring TCR-pMHC interfaces and demonstrate state-of-the-art results in recognizing binding-enhancing mutations.
HERMES: Holographic Equivariant neuRal network model for Mutational Effect and Stability prediction
Jul 09, 2024



Predicting the stability and fitness effects of amino acid mutations in proteins is a cornerstone of biological discovery and engineering. Various experimental techniques have been developed to measure mutational effects, providing us with extensive datasets across a diverse range of proteins. By training on these data, traditional computational modeling and more recent machine learning approaches have advanced significantly in predicting mutational effects. Here, we introduce HERMES, a 3D rotationally equivariant structure-based neural network model for mutational effect and stability prediction. Pre-trained to predict amino acid propensity from its surrounding 3D structure, HERMES can be fine-tuned for mutational effects using our open-source code. We present a suite of HERMES models, pre-trained with different strategies, and fine-tuned to predict the stability effect of mutations. Benchmarking against other models shows that HERMES often outperforms or matches their performance in predicting mutational effect on stability, binding, and fitness. HERMES offers versatile tools for evaluating mutational effects and can be fine-tuned for specific predictive objectives.
H-Packer: Holographic Rotationally Equivariant Convolutional Neural Network for Protein Side-Chain Packing
Nov 28, 2023



Accurately modeling protein 3D structure is essential for the design of functional proteins. An important sub-task of structure modeling is protein side-chain packing: predicting the conformation of side-chains (rotamers) given the protein's backbone structure and amino-acid sequence. Conventional approaches for this task rely on expensive sampling procedures over hand-crafted energy functions and rotamer libraries. Recently, several deep learning methods have been developed to tackle the problem in a data-driven way, albeit with vastly different formulations (from image-to-image translation to directly predicting atomic coordinates). Here, we frame the problem as a joint regression over the side-chains' true degrees of freedom: the dihedral $\chi$ angles. We carefully study possible objective functions for this task, while accounting for the underlying symmetries of the task. We propose Holographic Packer (H-Packer), a novel two-stage algorithm for side-chain packing built on top of two light-weight rotationally equivariant neural networks. We evaluate our method on CASP13 and CASP14 targets. H-Packer is computationally efficient and shows favorable performance against conventional physics-based algorithms and is competitive against alternative deep learning solutions.
Holographic-AE: an end-to-end SO-Equivariant Autoencoder in Fourier Space
Sep 30, 2022Group-equivariant neural networks have emerged as a data-efficient approach to solve classification and regression tasks, while respecting the relevant symmetries of the data. However, little work has been done to extend this paradigm to the unsupervised and generative domains. Here, we present Holographic-(V)AE (H-(V)AE), a fully end-to-end SO(3)-equivariant (variational) autoencoder in Fourier space, suitable for unsupervised learning and generation of data distributed around a specified origin. H-(V)AE is trained to reconstruct the spherical Fourier encoding of data, learning in the process a latent space with a maximally informative invariant embedding alongside an equivariant frame describing the orientation of the data. We extensively test the performance of H-(V)AE on diverse datasets and show that its latent space efficiently encodes the categorical features of spherical images and structural features of protein atomic environments. Our work can further be seen as a case study for equivariant modeling of a data distribution by reconstructing its Fourier encoding.
Approximate Bayesian Computation for an Explicit-Duration Hidden Markov Model of COVID-19 Hospital Trajectories
Apr 28, 2021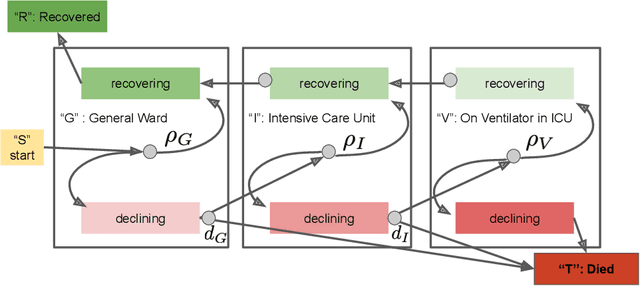
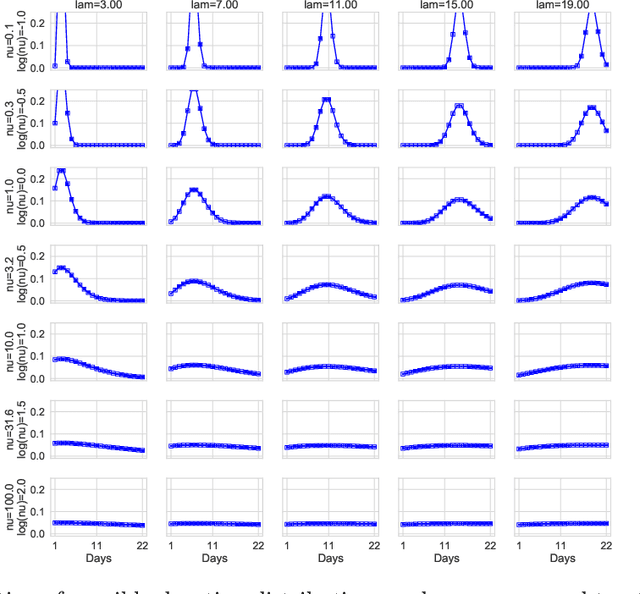
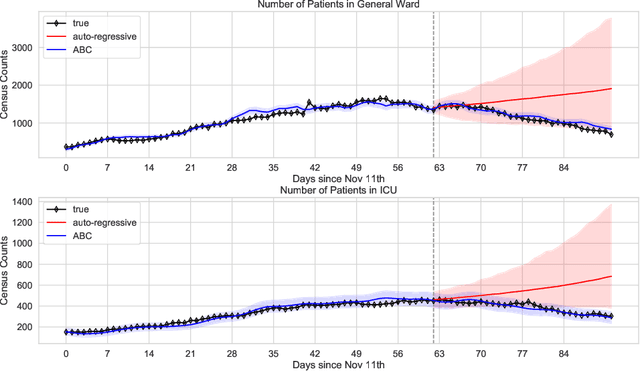
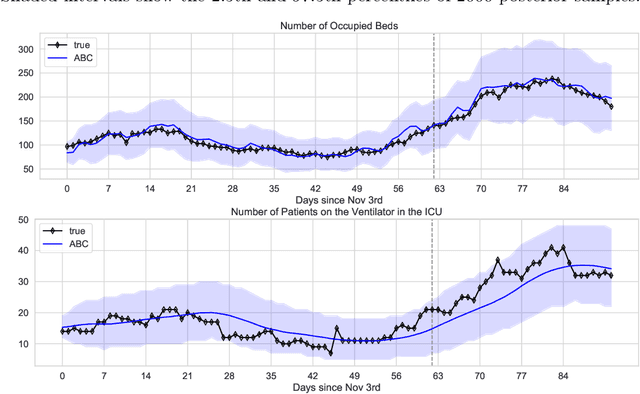
We address the problem of modeling constrained hospital resources in the midst of the COVID-19 pandemic in order to inform decision-makers of future demand and assess the societal value of possible interventions. For broad applicability, we focus on the common yet challenging scenario where patient-level data for a region of interest are not available. Instead, given daily admissions counts, we model aggregated counts of observed resource use, such as the number of patients in the general ward, in the intensive care unit, or on a ventilator. In order to explain how individual patient trajectories produce these counts, we propose an aggregate count explicit-duration hidden Markov model, nicknamed the ACED-HMM, with an interpretable, compact parameterization. We develop an Approximate Bayesian Computation approach that draws samples from the posterior distribution over the model's transition and duration parameters given aggregate counts from a specific location, thus adapting the model to a region or individual hospital site of interest. Samples from this posterior can then be used to produce future forecasts of any counts of interest. Using data from the United States and the United Kingdom, we show our mechanistic approach provides competitive probabilistic forecasts for the future even as the dynamics of the pandemic shift. Furthermore, we show how our model provides insight about recovery probabilities or length of stay distributions, and we suggest its potential to answer challenging what-if questions about the societal value of possible interventions.
Hierarchical Classification of Enzyme Promiscuity Using Positive, Unlabeled, and Hard Negative Examples
Feb 18, 2020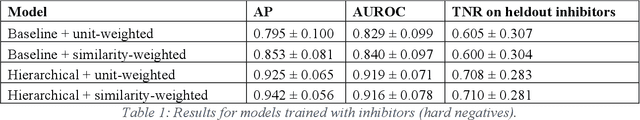
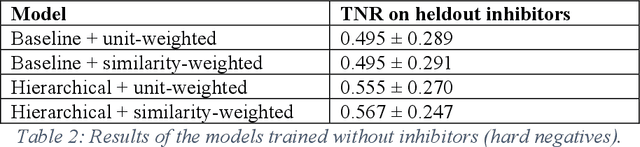
Despite significant progress in sequencing technology, there are many cellular enzymatic activities that remain unknown. We develop a new method, referred to as SUNDRY (Similarity-weighting for UNlabeled Data in a Residual HierarchY), for training enzyme-specific predictors that take as input a query substrate molecule and return whether the enzyme would act on that substrate or not. When addressing this enzyme promiscuity prediction problem, a major challenge is the lack of abundant labeled data, especially the shortage of labeled data for negative cases (enzyme-substrate pairs where the enzyme does not act to transform the substrate to a product molecule). To overcome this issue, our proposed method can learn to classify a target enzyme by sharing information from related enzymes via known tree hierarchies. Our method can also incorporate three types of data: those molecules known to be catalyzed by an enzyme (positive cases), those with unknown relationships (unlabeled cases), and molecules labeled as inhibitors for the enzyme. We refer to inhibitors as hard negative cases because they may be difficult to classify well: they bind to the enzyme, like positive cases, but are not transformed by the enzyme. Our method uses confidence scores derived from structural similarity to treat unlabeled examples as weighted negatives. We compare our proposed hierarchy-aware predictor against a baseline that cannot share information across related enzymes. Using data from the BRENDA database, we show that each of our contributions (hierarchical sharing, per-example confidence weighting of unlabeled data based on molecular similarity, and including inhibitors as hard-negative examples) contributes towards a better characterization of enzyme promiscuity.