 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhyloVAE: Unsupervised Learning of Phylogenetic Trees via Variational Autoencoders
Feb 07, 2025Learning informative representations of phylogenetic tree structures is essential for analyzing evolutionary relationships. Classical distance-based methods have been widely used to project phylogenetic trees into Euclidean space, but they are often sensitive to the choice of distance metric and may lack sufficient resolution. In this paper, we introduce phylogenetic variational autoencoders (PhyloVAEs), an unsupervised learning framework designed for representation learning and generative modeling of tree topologies. Leveraging an efficient encoding mechanism inspired by autoregressive tree topology generation, we develop a deep latent-variable generative model that facilitates fast, parallelized topology generation. PhyloVAE combines this generative model with a collaborative inference model based on learnable topological features, allowing for high-resolution representations of phylogenetic tree samples. Extensive experiments demonstrate PhyloVAE's robust representation learning capabilities and fast generation of phylogenetic tree topologies.
Variational Bayesian Phylogenetic Inference with Semi-implicit Branch Length Distributions
Aug 09, 2024Reconstructing the evolutionary history relating a collection of molecular sequences is the main subject of modern Bayesian phylogenetic inference. However, the commonly used Markov chain Monte Carlo methods can be inefficient due to the complicated space of phylogenetic trees, especially when the number of sequences is large. An alternative approach is variational Bayesian phylogenetic inference (VBPI) which transforms the inference problem into an optimization problem. While effective, the default diagonal lognormal approximation for the branch lengths of the tree used in VBPI is often insufficient to capture the complexity of the exact posterior. In this work, we propose a more flexible family of branch length variational posteriors based on semi-implicit hierarchical distributions using graph neural networks. We show that this semi-implicit construction emits straightforward permutation equivariant distributions, and therefore can handle the non-Euclidean branch length space across different tree topologies with ease. To deal with the intractable marginal probability of semi-implicit variational distributions, we develop several alternative lower bounds for stochastic optimization. We demonstrate the effectiveness of our proposed method over baseline methods on benchmark data examples, in terms of both marginal likelihood estimation and branch length posterior approximation.
A Variational Approach to Bayesian Phylogenetic Inference
Apr 16, 2022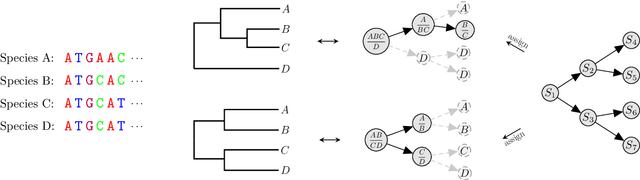
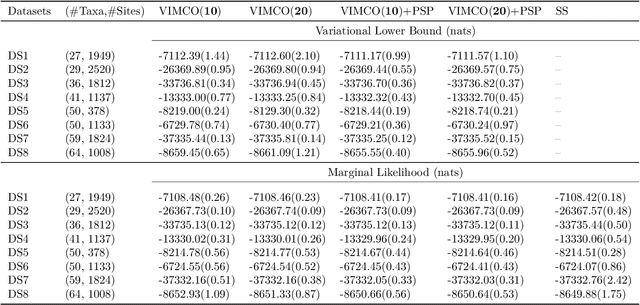
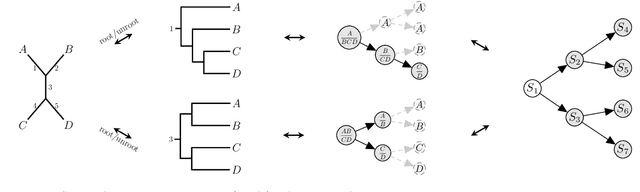
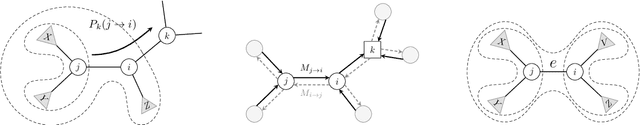
Bayesian phylogenetic inference is currently done via Markov chain Monte Carlo (MCMC) with simple proposal mechanisms. This hinders exploration efficiency and often requires long runs to deliver accurate posterior estimates. In this paper, we present an alternative approach: a variational framework for Bayesian phylogenetic analysis. We propose combining subsplit Bayesian networks, an expressive graphical model for tree topology distributions, and a structured amortization of the branch lengths over tree topologies for a suitable variational family of distributions. We train the variational approximation via stochastic gradient ascent and adopt gradient estimators for continuous and discrete variational parameters separately to deal with the composite latent space of phylogenetic models. We show that our variational approach provides competitive performance to MCMC, while requiring much less computation due to a more efficient exploration mechanism enabled by variational inference. Experiments on a benchmark of challenging real data Bayesian phylogenetic inference problems demonstrate the effectiveness and efficiency of our methods.
Non-bifurcating phylogenetic tree inference via the adaptive LASSO
May 28, 2018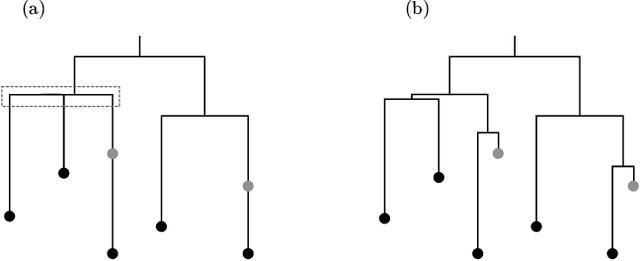
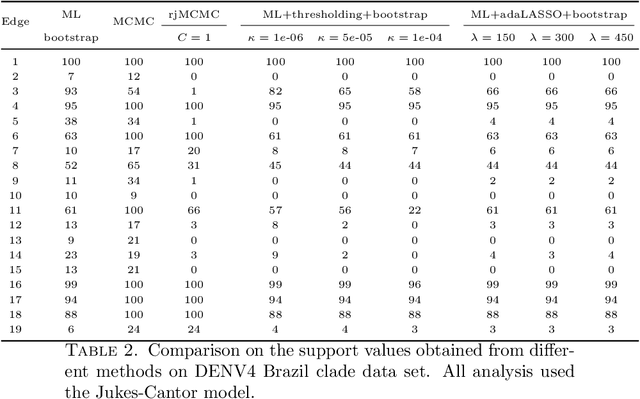
Phylogenetic tree inference using deep DNA sequencing is reshaping our understanding of rapidly evolving systems, such as the within-host battle between viruses and the immune system. Densely sampled phylogenetic trees can contain special features, including "sampled ancestors" in which we sequence a genotype along with its direct descendants, and "polytomies" in which multiple descendants arise simultaneously. These features are apparent after identifying zero-length branches in the tree. However, current maximum-likelihood based approaches are not capable of revealing such zero-length branches. In this paper, we find these zero-length branches by introducing adaptive-LASSO-type regularization estimators to phylogenetics, deriving their properties, and showing regularization to be a practically useful approach for phylogenetics.