 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Perspective on AI-Guided Molecular Simulations in VR: Exploring Strategies for Imitation Learning in Hyperdimensional Molecular Systems
Sep 11, 2024Molecular dynamics simulations are a crucial computational tool for researchers to understand and engineer molecular structure and function in areas such as drug discovery, protein engineering, and material design. Despite their utility, MD simulations are expensive, owing to the high dimensionality of molecular systems. Interactive molecular dynamics in virtual reality (iMD-VR) has recently been developed as a 'human-in-the-loop' strategy, which leverages high-performance computing to accelerate the researcher's ability to solve the hyperdimensional sampling problem. By providing an immersive 3D environment that enables visualization and manipulation of real-time molecular motion, iMD-VR enables researchers and students to efficiently and intuitively explore and navigate these complex, high-dimensional systems. iMD-VR platforms offer a unique opportunity to quickly generate rich datasets that capture human experts' spatial insight regarding molecular structure and function. This paper explores the possibility of employing user-generated iMD-VR datasets to train AI agents via imitation learning (IL). IL is an important technique in robotics that enables agents to mimic complex behaviors from expert demonstrations, thus circumventing the need for explicit programming or intricate reward design. We review the utilization of IL for manipulation tasks in robotics and discuss how iMD-VR recordings could be used to train IL models for solving specific molecular 'tasks'. We then investigate how such approaches could be applied to the data captured from iMD-VR recordings. Finally, we outline the future research directions and potential challenges of using AI agents to augment human expertise to efficiently navigate conformational spaces, highlighting how this approach could provide valuable insight across domains such as materials science, protein engineering, and computer-aided drug design.
A community-powered search of machine learning strategy space to find NMR property prediction models
Aug 13, 2020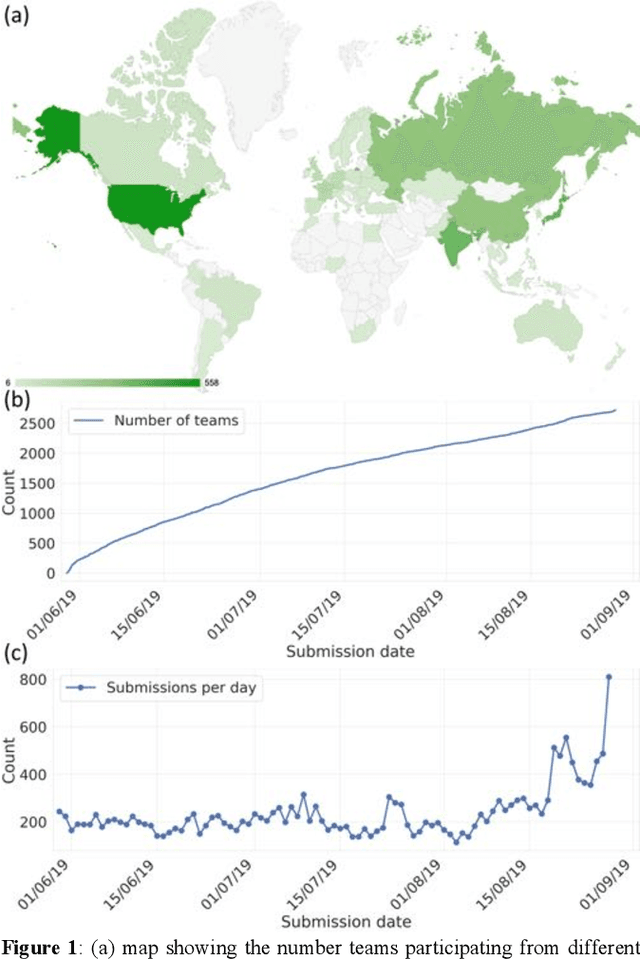
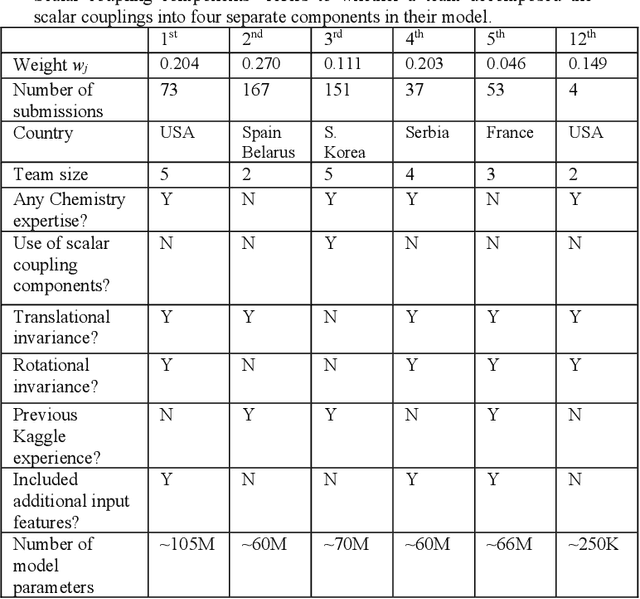
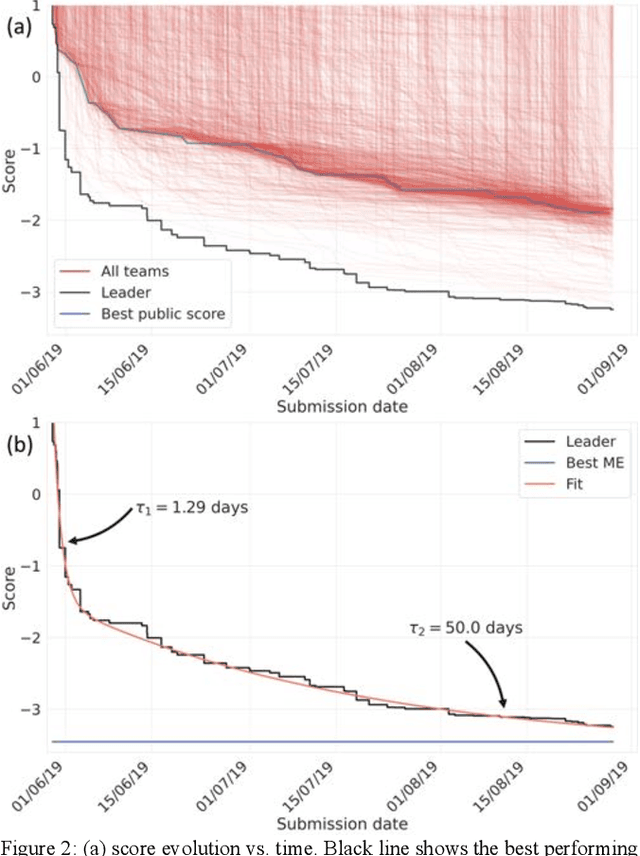
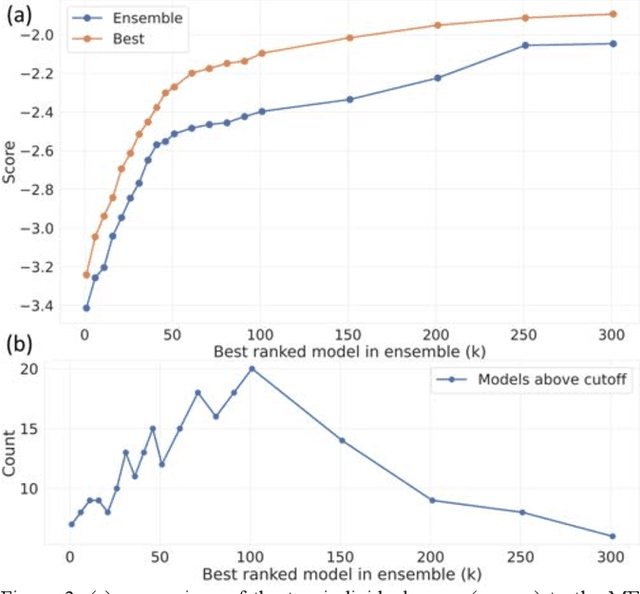
The rise of machine learning (ML) has created an explosion in the potential strategies for using data to make scientific predictions. For physical scientists wishing to apply ML strategies to a particular domain, it can be difficult to assess in advance what strategy to adopt within a vast space of possibilities. Here we outline the results of an online community-powered effort to swarm search the space of ML strategies and develop algorithms for predicting atomic-pairwise nuclear magnetic resonance (NMR) properties in molecules. Using an open-source dataset, we worked with Kaggle to design and host a 3-month competition which received 47,800 ML model predictions from 2,700 teams in 84 countries. Within 3 weeks, the Kaggle community produced models with comparable accuracy to our best previously published "in-house" efforts. A meta-ensemble model constructed as a linear combination of the top predictions has a prediction accuracy which exceeds that of any individual model, 7-19x better than our previous state-of-the-art. The results highlight the potential of transformer architectures for predicting quantum mechanical (QM) molecular properties.