 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePOPSICLE: Benchmark Datasets for Segmentation and Localization in CryoET
Jun 08, 2026Cryo-electron tomography (cryoET) has emerged as a powerful tool in structural and cellular biology by enabling direct visualization of macromolecular structures within intact cells, thereby linking molecular architecture to cellular organization in a native context. Realizing the full potential of cryoET, however, increasingly depends on advances in computational analysis, particularly machine learning (ML), to interpret its complex and information-rich data. Despite rapid progress, ML development for cryoET remains bottlenecked by the lack of standardized, well-annotated benchmarks. Existing evaluations are typically small, task-specific, and are assembled in isolation, limiting robust comparisons across methods. Here, we present POPSICLE, a benchmark suite for cryoET segmentation and macromolecular localization built from the CryoET Data Portal - an open, ML-ready repository of tomographic data, metadata, and annotations. POPSICLE spans eukaryotic and prokaryotic systems, both purified and fully in situ samples, and dense voxel-wise segmentation as well as sparse localization tasks. Built on a living data resource, it can expand as new datasets and annotations become available. Baseline experiments reveal substantial variation in model rankings across tasks, underscoring the need for benchmarks tailored to the unique characteristics of cryoET rather than evaluation practices adapted from adjacent biomedical imaging domains. POPSICLE thus provides an open and extensible foundation for reproducible ML evaluation in cryoET.
ModelAngelo: Automated Model Building in Cryo-EM Maps
Sep 30, 2022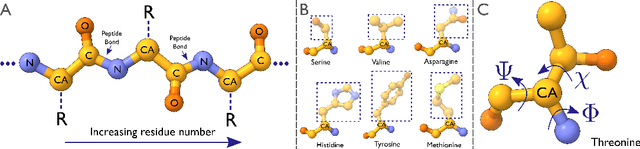
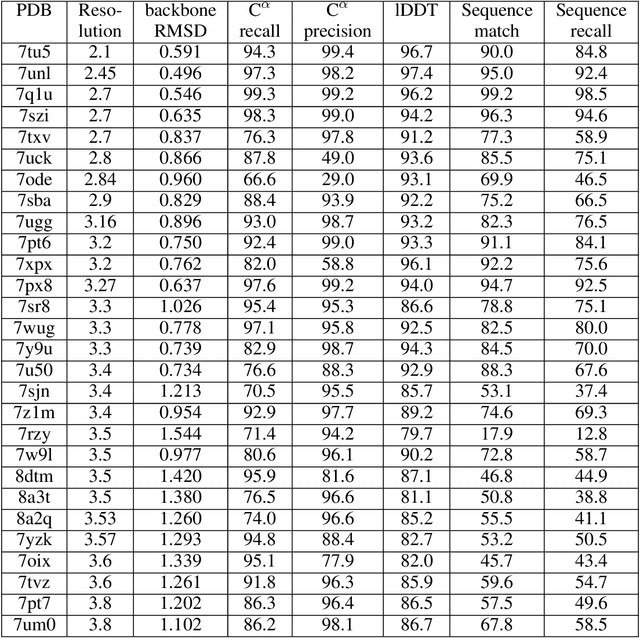
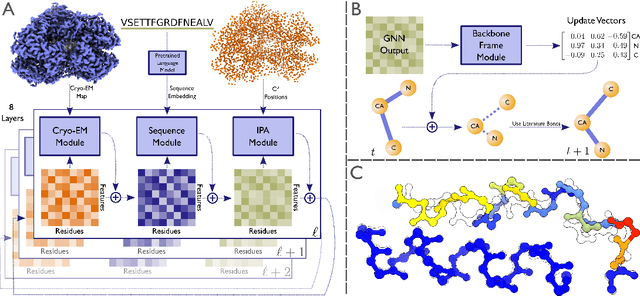
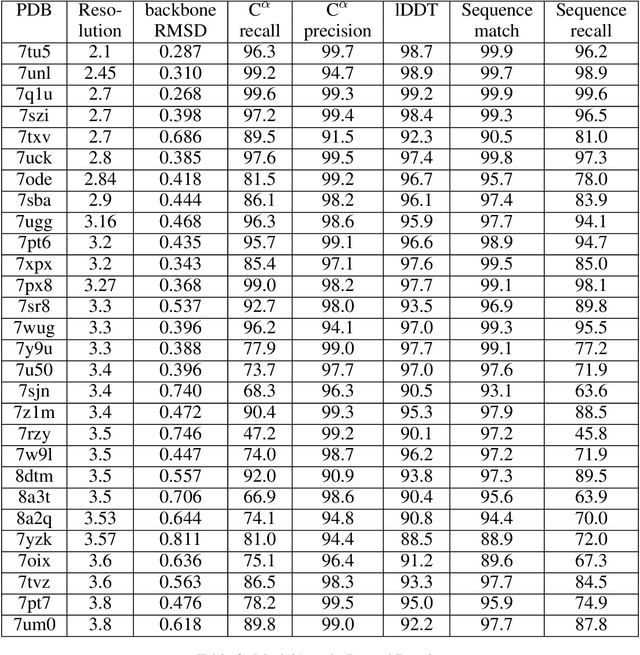
Electron cryo-microscopy (cryo-EM) produces three-dimensional (3D) maps of the electrostatic potential of biological macromolecules, including proteins. At sufficient resolution, the cryo-EM maps, along with some knowledge about the imaged molecules, allow de novo atomic modelling. Typically, this is done through a laborious manual process. Recent advances in machine learning applications to protein structure prediction show potential for automating this process. Taking inspiration from these techniques, we have built ModelAngelo for automated model building of proteins in cryo-EM maps. ModelAngelo first uses a residual convolutional neural network (CNN) to initialize a graph representation with nodes assigned to individual amino acids of the proteins in the map and edges representing the protein chain. The graph is then refined with a graph neural network (GNN) that combines the cryo-EM data, the amino acid sequence data and prior knowledge about protein geometries. The GNN refines the geometry of the protein chain and classifies the amino acids for each of its nodes. The final graph is post-processed with a hidden Markov model (HMM) search to map each protein chain to entries in a user provided sequence file. Application to 28 test cases shows that ModelAngelo outperforms the state-of-the-art and approximates manual building for cryo-EM maps with resolutions better than 3.5 \r{A}.