 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning of coarse-grained Molecular Dynamics Force Fields
Dec 08, 2018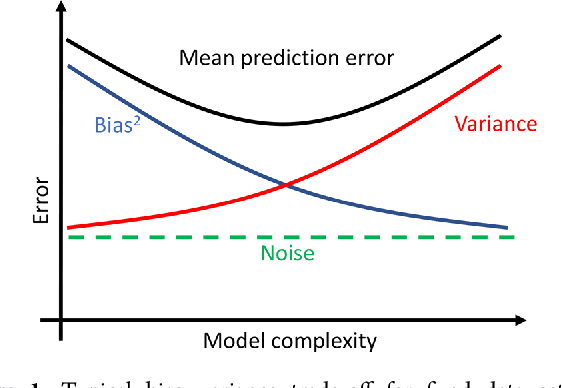
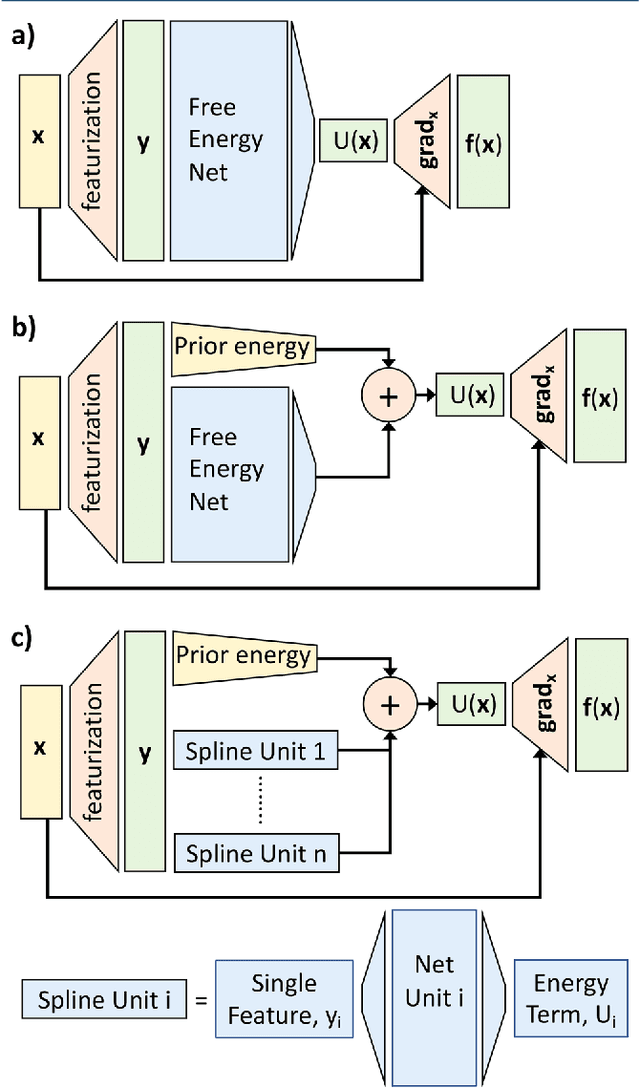
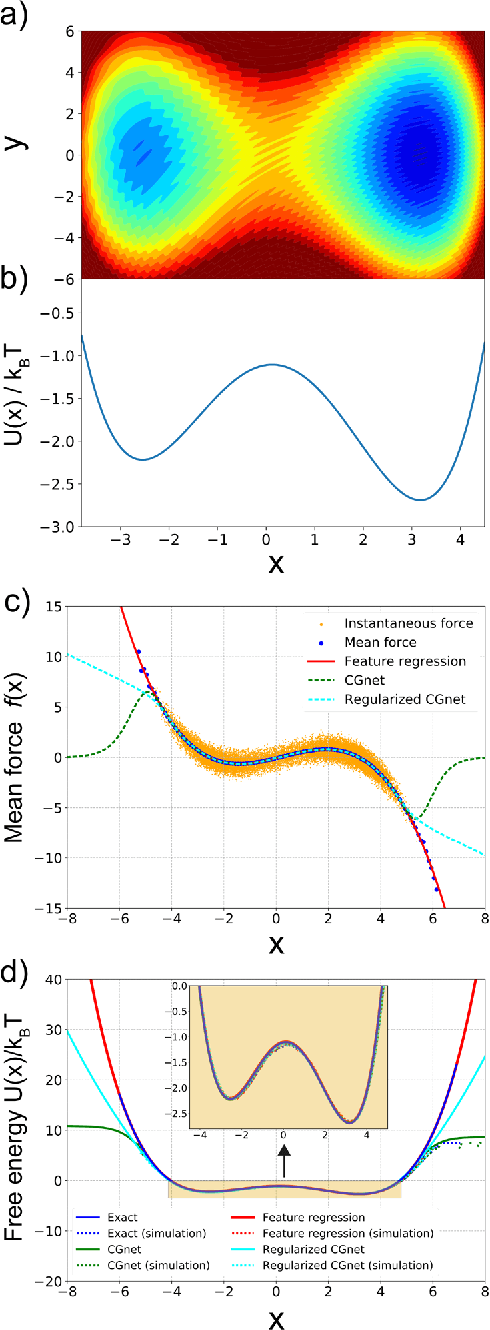
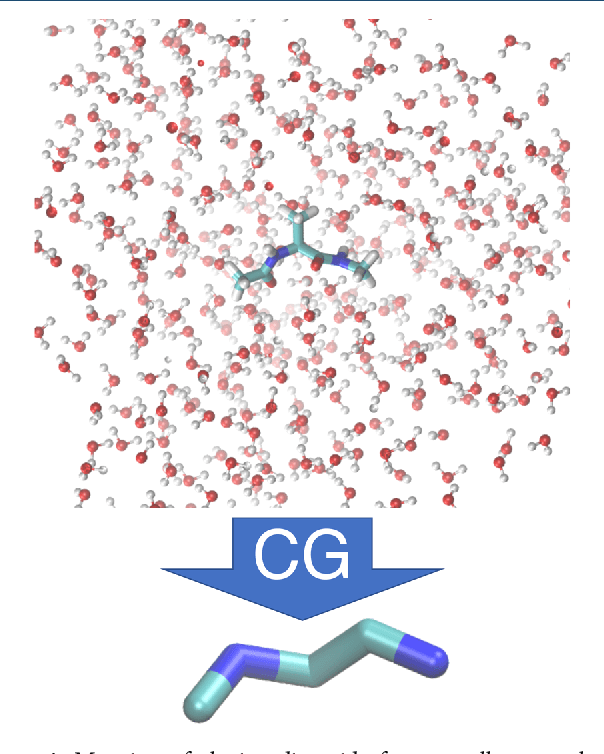
Atomistic or ab-initio molecular dynamics simulations are widely used to predict thermodynamics and kinetics and relate them to molecular structure. A common approach to go beyond the time- and lengthscales accessible with such computationally expensive simulations is the definition of coarse-grained molecular models. Existing coarse-graining approaches define an effective interaction potential to match defined properties of high-resolution models or experimental data. In this paper we reformulate coarse-graining as a supervised machine learning problem. We use statistical learning theory to decompose the coarse-graining error and cross-validation to select to compare the performance of different models. We introduce CGnets, a deep learning approach, that learn coarse-grained free energy functions and can be trained by the force matching scheme. CGnets maintain all physically relevant invariances and allow to incorporate prior physics knowledge to avoid sampling of unphysical structures. We demonstrate that CGnets outperform the results of classical coarse-graining methods, as they are able to capture the multi-body terms that emerge from the dimensionality reduction.
Time-lagged autoencoders: Deep learning of slow collective variables for molecular kinetics
Oct 30, 2017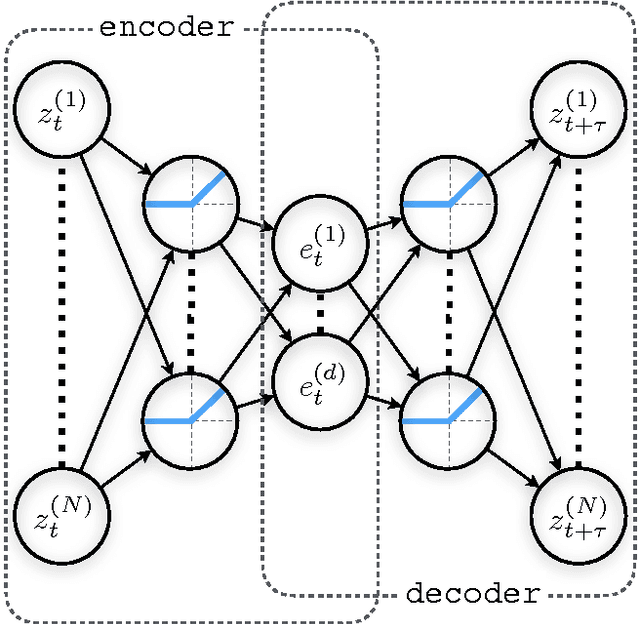
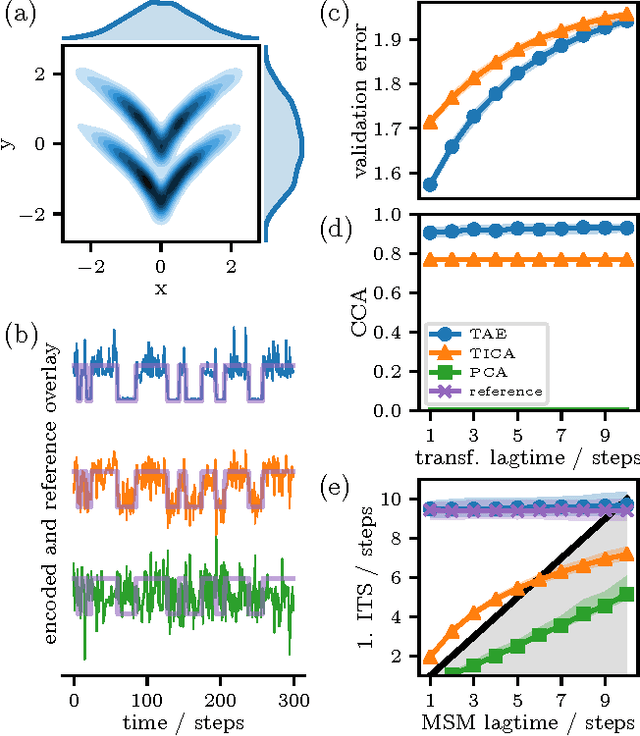
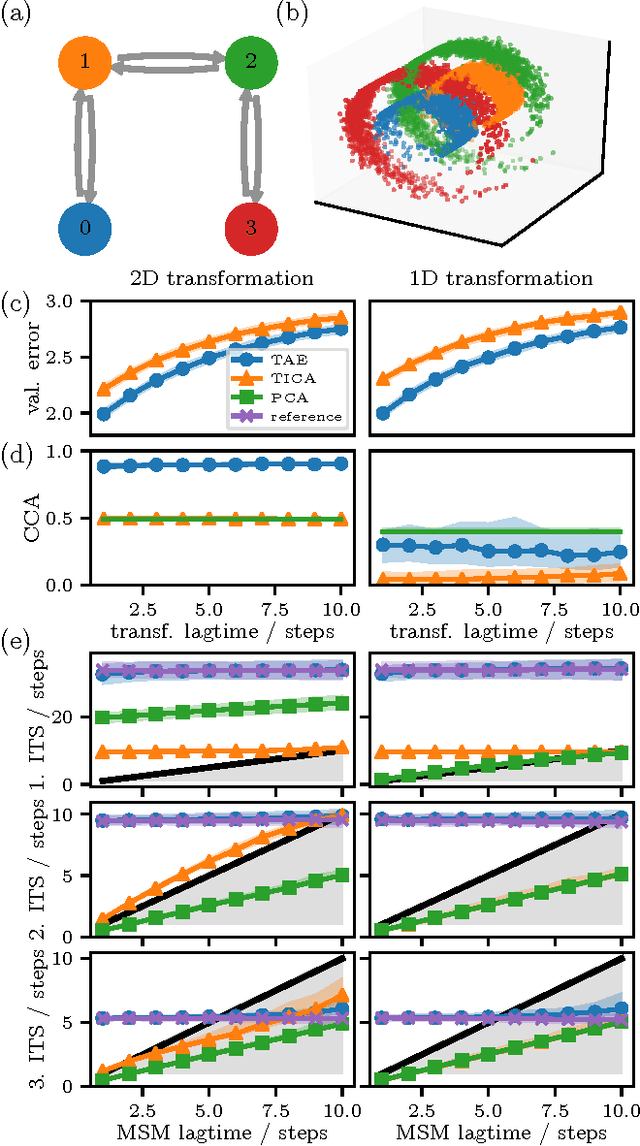
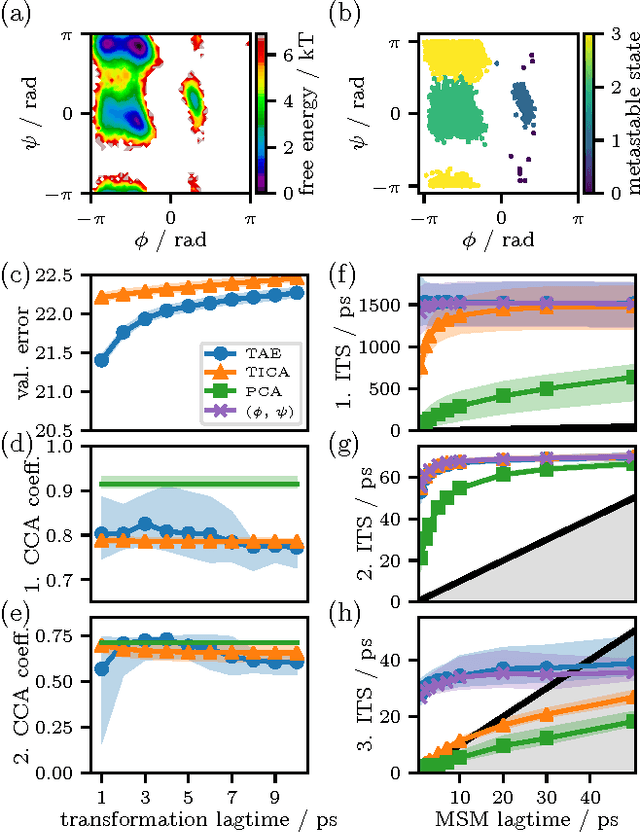
Inspired by the success of deep learning techniques in the physical and chemical sciences, we apply a modification of an autoencoder type deep neural network to the task of dimension reduction of molecular dynamics data. We can show that our time-lagged autoencoder reliably finds low-dimensional embeddings for high-dimensional feature spaces which capture the slow dynamics of the underlying stochastic processes - beyond the capabilities of linear dimension reduction techniques.