 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeutron and X-ray Diffraction Reveal the Limits of Long-Range Machine Learning Potentials for Medium-Range Order in Silica Glass
Apr 23, 2026Glassy silica is a foundational material in optics and electronics, yet accurately predicting its medium-range order (MRO) remains a major challenge for machine-learning interatomic potentials (MLIPs). While local MLIPs reproduce the short-range SiO4 tetrahedral network well, it remains unclear whether locality alone is sufficient to recover the first sharp diffraction peak (FSDP), the principal experimental signature of MRO. Here, we combine neutron and X-ray diffraction measurements with large-scale molecular dynamics driven by two MACE-based models: a short-range (SR) potential and a long-range (LR) extension incorporating reciprocal-space gated attention. The SR model systematically over-structures the network, producing an overly intense FSDP in both the liquid and glassy states. Incorporating long-range interactions improves agreement with experiment for the liquid structure by reducing this excess ordering, but the LR model still fails to recover the experimental amorphous MRO after quenching. Ring-statistics and bond-angle analyses reveal that SR model exhibits an artificially narrow distribution dominated by six-membered rings, while the LR model produces a broader but still biased ring population. Despite preserving the correct tetrahedral geometry, both models show limited variability in Si-O-Si angles, indicating constrained network flexibility. These structural signatures demonstrate that both models retain excessive memory of the parent liquid network, leading to kinetically trapped and nonphysical medium-range configurations during vitrification. These results show that explicit long-range interactions are necessary but not sufficient for predictive modelling of disordered silica and suggest that accurate MRO further requires training data and sampling strategies that adequately represent the liquid-to-glass transition.
Machine Learning Inter-Atomic Potentials Generation Driven by Active Learning: A Case Study for Amorphous and Liquid Hafnium dioxide
Oct 22, 2019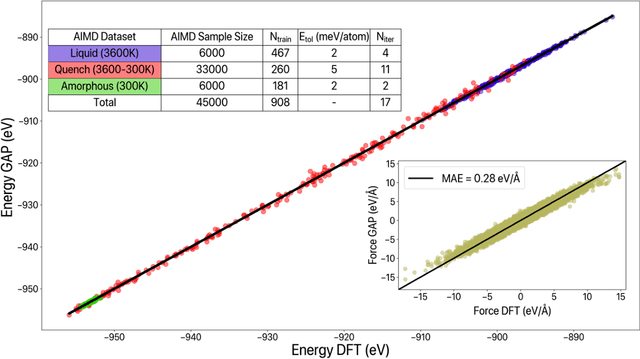
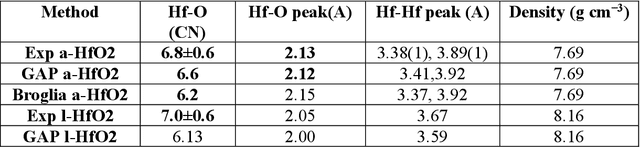
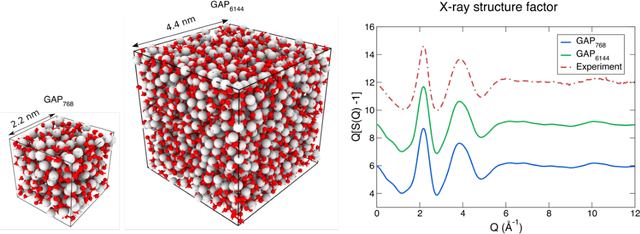
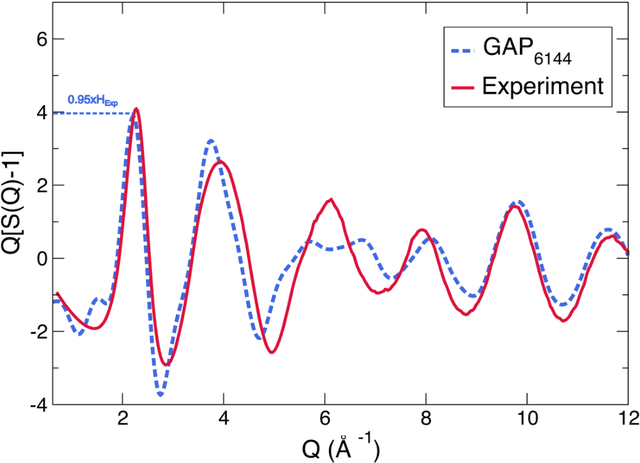
We propose a novel active learning scheme for automatically sampling a minimum number of uncorrelated configurations for fitting the Gaussian Approximation Potential (GAP). Our active learning scheme consists of an unsupervised machine learning (ML) scheme coupled to Bayesian optimization technique that evaluates the GAP model. We apply this scheme to a Hafnium dioxide (HfO2) dataset generated from a melt-quench ab initio molecular dynamics (AIMD) protocol. Our results show that the active learning scheme, with no prior knowledge of the dataset is able to extract a configuration that reaches the required energy fit tolerance. Further, molecular dynamics (MD) simulations performed using this active learned GAP model on 6144-atom systems of amorphous and liquid state elucidate the structural properties of HfO2 with near ab initio precision and quench rates (i.e. 1.0 K/ps) not accessible via AIMD. The melt and amorphous x-ray structural factors generated from our simulation are in good agreement with experiment. Additionally, the calculated diffusion constants are in good agreement with previous ab initio studies.