 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTopological Signatures of Adversaries in Multimodal Alignments
Jan 29, 2025Multimodal Machine Learning systems, particularly those aligning text and image data like CLIP/BLIP models, have become increasingly prevalent, yet remain susceptible to adversarial attacks. While substantial research has addressed adversarial robustness in unimodal contexts, defense strategies for multimodal systems are underexplored. This work investigates the topological signatures that arise between image and text embeddings and shows how adversarial attacks disrupt their alignment, introducing distinctive signatures. We specifically leverage persistent homology and introduce two novel Topological-Contrastive losses based on Total Persistence and Multi-scale kernel methods to analyze the topological signatures introduced by adversarial perturbations. We observe a pattern of monotonic changes in the proposed topological losses emerging in a wide range of attacks on image-text alignments, as more adversarial samples are introduced in the data. By designing an algorithm to back-propagate these signatures to input samples, we are able to integrate these signatures into Maximum Mean Discrepancy tests, creating a novel class of tests that leverage topological signatures for better adversarial detection.
LoRID: Low-Rank Iterative Diffusion for Adversarial Purification
Sep 12, 2024



This work presents an information-theoretic examination of diffusion-based purification methods, the state-of-the-art adversarial defenses that utilize diffusion models to remove malicious perturbations in adversarial examples. By theoretically characterizing the inherent purification errors associated with the Markov-based diffusion purifications, we introduce LoRID, a novel Low-Rank Iterative Diffusion purification method designed to remove adversarial perturbation with low intrinsic purification errors. LoRID centers around a multi-stage purification process that leverages multiple rounds of diffusion-denoising loops at the early time-steps of the diffusion models, and the integration of Tucker decomposition, an extension of matrix factorization, to remove adversarial noise at high-noise regimes. Consequently, LoRID increases the effective diffusion time-steps and overcomes strong adversarial attacks, achieving superior robustness performance in CIFAR-10/100, CelebA-HQ, and ImageNet datasets under both white-box and black-box settings.
LaFA: Latent Feature Attacks on Non-negative Matrix Factorization
Aug 07, 2024



As Machine Learning (ML) applications rapidly grow, concerns about adversarial attacks compromising their reliability have gained significant attention. One unsupervised ML method known for its resilience to such attacks is Non-negative Matrix Factorization (NMF), an algorithm that decomposes input data into lower-dimensional latent features. However, the introduction of powerful computational tools such as Pytorch enables the computation of gradients of the latent features with respect to the original data, raising concerns about NMF's reliability. Interestingly, naively deriving the adversarial loss for NMF as in the case of ML would result in the reconstruction loss, which can be shown theoretically to be an ineffective attacking objective. In this work, we introduce a novel class of attacks in NMF termed Latent Feature Attacks (LaFA), which aim to manipulate the latent features produced by the NMF process. Our method utilizes the Feature Error (FE) loss directly on the latent features. By employing FE loss, we generate perturbations in the original data that significantly affect the extracted latent features, revealing vulnerabilities akin to those found in other ML techniques. To handle large peak-memory overhead from gradient back-propagation in FE attacks, we develop a method based on implicit differentiation which enables their scaling to larger datasets. We validate NMF vulnerabilities and FE attacks effectiveness through extensive experiments on synthetic and real-world data.
Robust Adversarial Defense by Tensor Factorization
Sep 03, 2023



As machine learning techniques become increasingly prevalent in data analysis, the threat of adversarial attacks has surged, necessitating robust defense mechanisms. Among these defenses, methods exploiting low-rank approximations for input data preprocessing and neural network (NN) parameter factorization have shown potential. Our work advances this field further by integrating the tensorization of input data with low-rank decomposition and tensorization of NN parameters to enhance adversarial defense. The proposed approach demonstrates significant defense capabilities, maintaining robust accuracy even when subjected to the strongest known auto-attacks. Evaluations against leading-edge robust performance benchmarks reveal that our results not only hold their ground against the best defensive methods available but also exceed all current defense strategies that rely on tensor factorizations. This study underscores the potential of integrating tensorization and low-rank decomposition as a robust defense against adversarial attacks in machine learning.
Less is more: sampling chemical space with active learning
Apr 09, 2018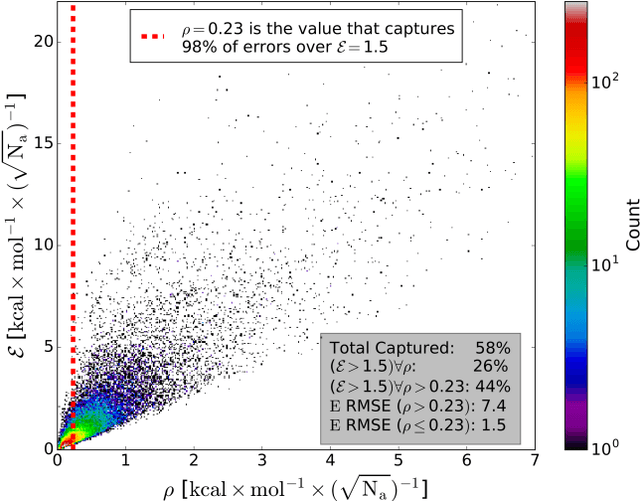
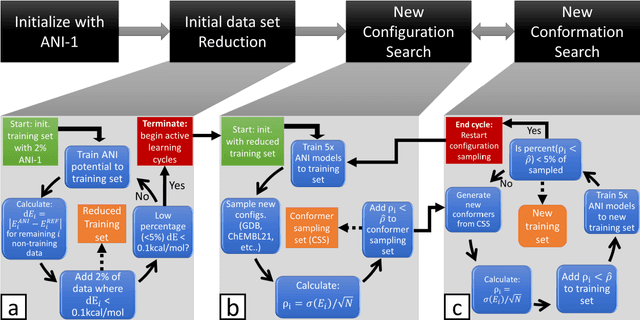
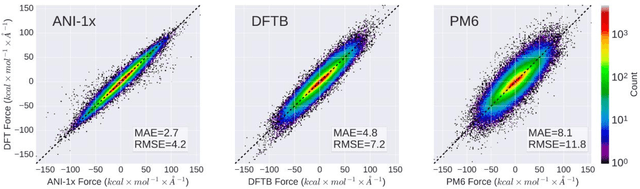
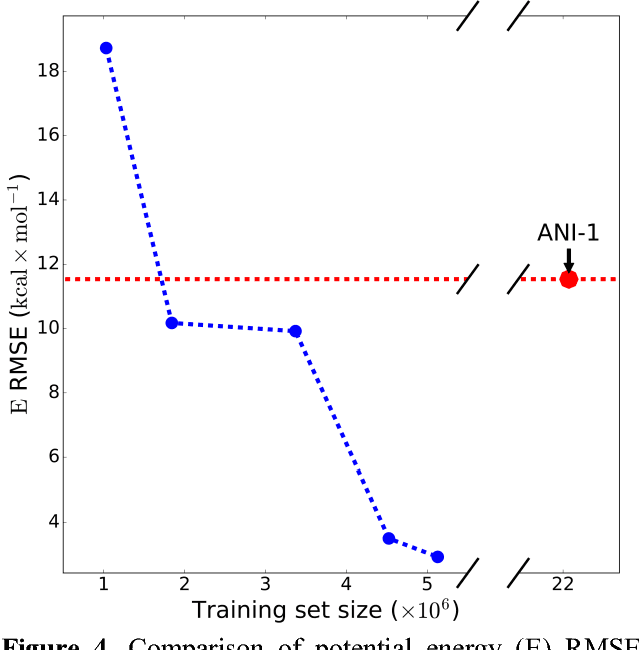
The development of accurate and transferable machine learning (ML) potentials for predicting molecular energetics is a challenging task. The process of data generation to train such ML potentials is a task neither well understood nor researched in detail. In this work, we present a fully automated approach for the generation of datasets with the intent of training universal ML potentials. It is based on the concept of active learning (AL) via Query by Committee (QBC), which uses the disagreement between an ensemble of ML potentials to infer the reliability of the ensemble's prediction. QBC allows the presented AL algorithm to automatically sample regions of chemical space where the ML potential fails to accurately predict the potential energy. AL improves the overall fitness of ANAKIN-ME (ANI) deep learning potentials in rigorous test cases by mitigating human biases in deciding what new training data to use. AL also reduces the training set size to a fraction of the data required when using naive random sampling techniques. To provide validation of our AL approach we develop the COMP6 benchmark (publicly available on GitHub), which contains a diverse set of organic molecules. Through the AL process, it is shown that the AL-based potentials perform as well as the ANI-1 potential on COMP6 with only 10% of the data, and vastly outperforms ANI-1 with 25% the amount of data. Finally, we show that our proposed AL technique develops a universal ANI potential (ANI-1x) that provides accurate energy and force predictions on the entire COMP6 benchmark. This universal ML potential achieves a level of accuracy on par with the best ML potentials for single molecule or materials, while remaining applicable to the general class of organic molecules comprised of the elements CHNO.
* Accepted at J. Chem. Phys