 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobust Node Affinities via Jaccard-Biased Random Walks and Rank Aggregation
Mar 05, 2026Estimating node similarity is a fundamental task in network analysis and graph-based machine learning, with applications in clustering, community detection, classification, and recommendation. We propose TopKGraphs, a method based on start-node-anchored random walks that bias transitions toward nodes with structurally similar neighborhoods, measured via Jaccard similarity. Rather than computing stationary distributions, walks are treated as stochastic neighborhood samplers, producing partial node rankings that are aggregated using robust rank aggregation to construct interpretable node-to-node affinity matrices. TopKGraphs provides a non-parametric, interpretable, and general-purpose representation of node similarity that can be applied in both network analysis and machine learning workflows. We evaluate the method on synthetic graphs (stochastic block models, Lancichinetti-Fortunato-Radicchi benchmark graphs), k-nearest-neighbor graphs from tabular datasets, and a curated high-confidence protein-protein interaction network. Across all scenarios, TopKGraphs achieves competitive or superior performance compared to standard similarity measures (Jaccard, Dice), a diffusion-based method (personalized PageRank), and an embedding-based approach (Node2Vec), demonstrating robustness in sparse, noisy, or heterogeneous networks. These results suggest that TopKGraphs is a versatile and interpretable tool for bridging simple local similarity measures with more complex embedding-based approaches, facilitating both data mining and network analysis applications.
Feature graphs for interpretable unsupervised tree ensembles: centrality, interaction, and application in disease subtyping
Apr 27, 2024Interpretable machine learning has emerged as central in leveraging artificial intelligence within high-stakes domains such as healthcare, where understanding the rationale behind model predictions is as critical as achieving high predictive accuracy. In this context, feature selection assumes a pivotal role in enhancing model interpretability by identifying the most important input features in black-box models. While random forests are frequently used in biomedicine for their remarkable performance on tabular datasets, the accuracy gained from aggregating decision trees comes at the expense of interpretability. Consequently, feature selection for enhancing interpretability in random forests has been extensively explored in supervised settings. However, its investigation in the unsupervised regime remains notably limited. To address this gap, the study introduces novel methods to construct feature graphs from unsupervised random forests and feature selection strategies to derive effective feature combinations from these graphs. Feature graphs are constructed for the entire dataset as well as individual clusters leveraging the parent-child node splits within the trees, such that feature centrality captures their relevance to the clustering task, while edge weights reflect the discriminating power of feature pairs. Graph-based feature selection methods are extensively evaluated on synthetic and benchmark datasets both in terms of their ability to reduce dimensionality while improving clustering performance, as well as to enhance model interpretability. An application on omics data for disease subtyping identifies the top features for each cluster, showcasing the potential of the proposed approach to enhance interpretability in clustering analyses and its utility in a real-world biomedical application.
Federated unsupervised random forest for privacy-preserving patient stratification
Jan 29, 2024In the realm of precision medicine, effective patient stratification and disease subtyping demand innovative methodologies tailored for multi-omics data. Clustering techniques applied to multi-omics data have become instrumental in identifying distinct subgroups of patients, enabling a finer-grained understanding of disease variability. This work establishes a powerful framework for advancing precision medicine through unsupervised random-forest-based clustering and federated computing. We introduce a novel multi-omics clustering approach utilizing unsupervised random-forests. The unsupervised nature of the random forest enables the determination of cluster-specific feature importance, unraveling key molecular contributors to distinct patient groups. Moreover, our methodology is designed for federated execution, a crucial aspect in the medical domain where privacy concerns are paramount. We have validated our approach on machine learning benchmark data sets as well as on cancer data from The Cancer Genome Atlas (TCGA). Our method is competitive with the state-of-the-art in terms of disease subtyping, but at the same time substantially improves the cluster interpretability. Experiments indicate that local clustering performance can be improved through federated computing.
Be Careful When Evaluating Explanations Regarding Ground Truth
Nov 08, 2023



Evaluating explanations of image classifiers regarding ground truth, e.g. segmentation masks defined by human perception, primarily evaluates the quality of the models under consideration rather than the explanation methods themselves. Driven by this observation, we propose a framework for $\textit{jointly}$ evaluating the robustness of safety-critical systems that $\textit{combine}$ a deep neural network with an explanation method. These are increasingly used in real-world applications like medical image analysis or robotics. We introduce a fine-tuning procedure to (mis)align model$\unicode{x2013}$explanation pipelines with ground truth and use it to quantify the potential discrepancy between worst and best-case scenarios of human alignment. Experiments across various model architectures and post-hoc local interpretation methods provide insights into the robustness of vision transformers and the overall vulnerability of such AI systems to potential adversarial attacks.
Explaining and visualizing black-box models through counterfactual paths
Aug 01, 2023Explainable AI (XAI) is an increasingly important area of machine learning research, which aims to make black-box models transparent and interpretable. In this paper, we propose a novel approach to XAI that uses the so-called counterfactual paths generated by conditional permutations of features. The algorithm measures feature importance by identifying sequential permutations of features that most influence changes in model predictions. It is particularly suitable for generating explanations based on counterfactual paths in knowledge graphs incorporating domain knowledge. Counterfactual paths introduce an additional graph dimension to current XAI methods in both explaining and visualizing black-box models. Experiments with synthetic and medical data demonstrate the practical applicability of our approach.
Bayesian post-hoc regularization of random forests
Jun 06, 2023



Random Forests are powerful ensemble learning algorithms widely used in various machine learning tasks. However, they have a tendency to overfit noisy or irrelevant features, which can result in decreased generalization performance. Post-hoc regularization techniques aim to mitigate this issue by modifying the structure of the learned ensemble after its training. Here, we propose Bayesian post-hoc regularization to leverage the reliable patterns captured by leaf nodes closer to the root, while potentially reducing the impact of more specific and potentially noisy leaf nodes deeper in the tree. This approach allows for a form of pruning that does not alter the general structure of the trees but rather adjusts the influence of leaf nodes based on their proximity to the root node. We have evaluated the performance of our method on various machine learning data sets. Our approach demonstrates competitive performance with the state-of-the-art methods and, in certain cases, surpasses them in terms of predictive accuracy and generalization.
Parea: multi-view ensemble clustering for cancer subtype discovery
Sep 30, 2022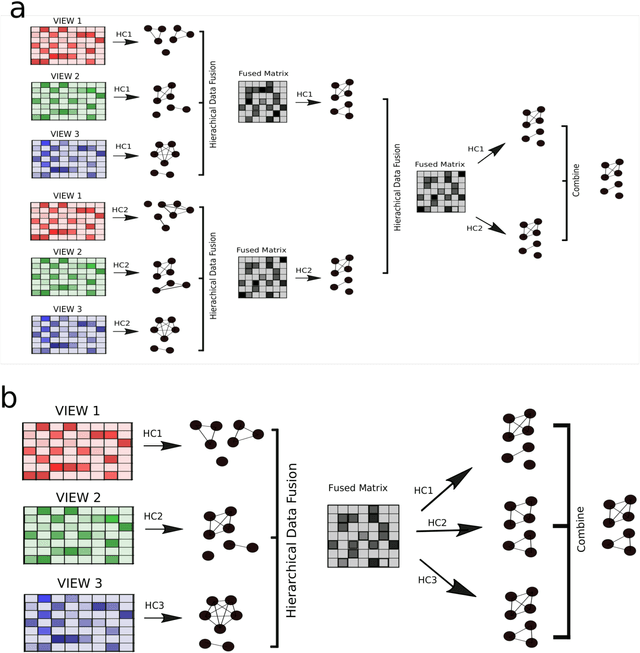
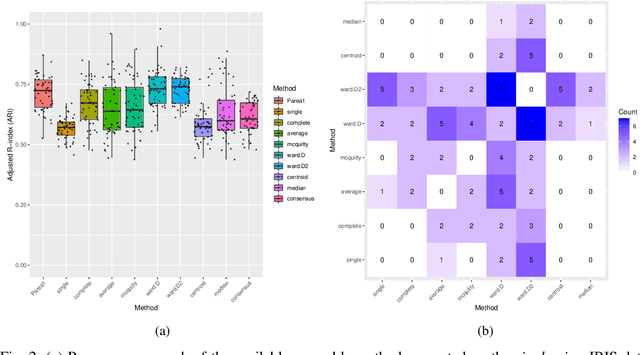
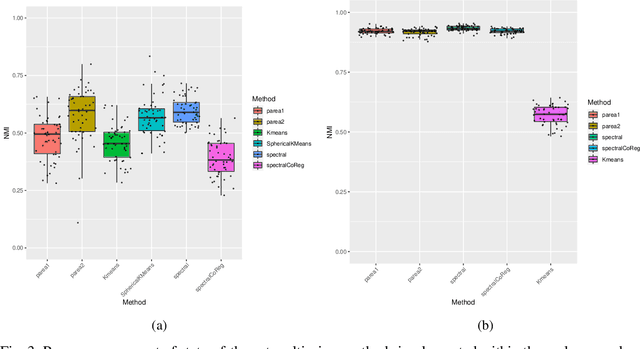
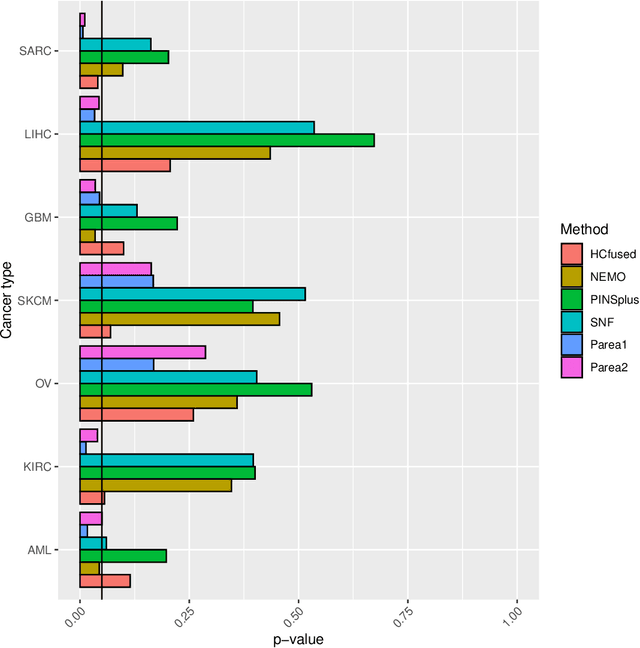
Multi-view clustering methods are essential for the stratification of patients into sub-groups of similar molecular characteristics. In recent years, a wide range of methods has been developed for this purpose. However, due to the high diversity of cancer-related data, a single method may not perform sufficiently well in all cases. We present Parea, a multi-view hierarchical ensemble clustering approach for disease subtype discovery. We demonstrate its performance on several machine learning benchmark datasets. We apply and validate our methodology on real-world multi-view cancer patient data. Parea outperforms the current state-of-the-art on six out of seven analysed cancer types. We have integrated the Parea method into our developed Python package Pyrea (https://github.com/mdbloice/Pyrea), which enables the effortless and flexible design of ensemble workflows while incorporating a wide range of fusion and clustering algorithms.
Network Module Detection from Multi-Modal Node Features with a Greedy Decision Forest for Actionable Explainable AI
Aug 26, 2021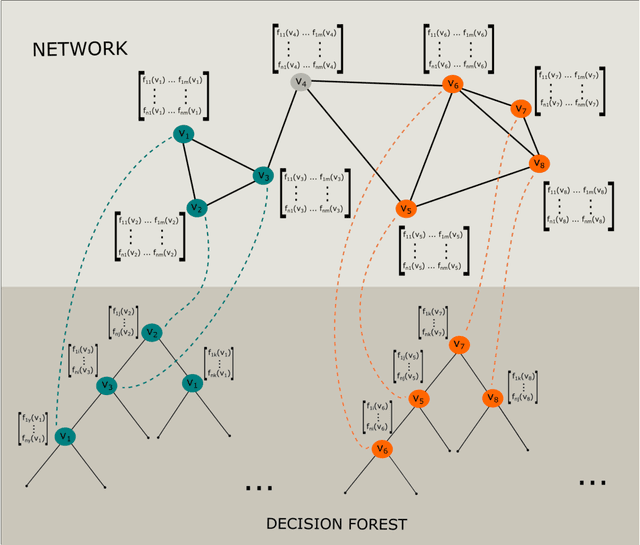
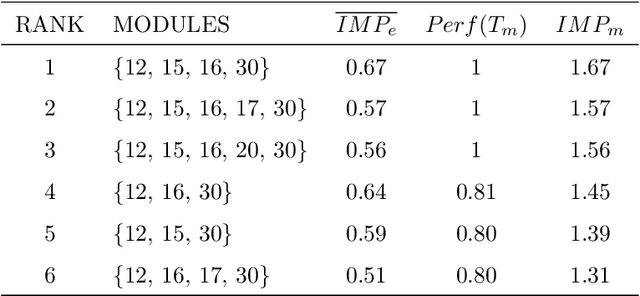
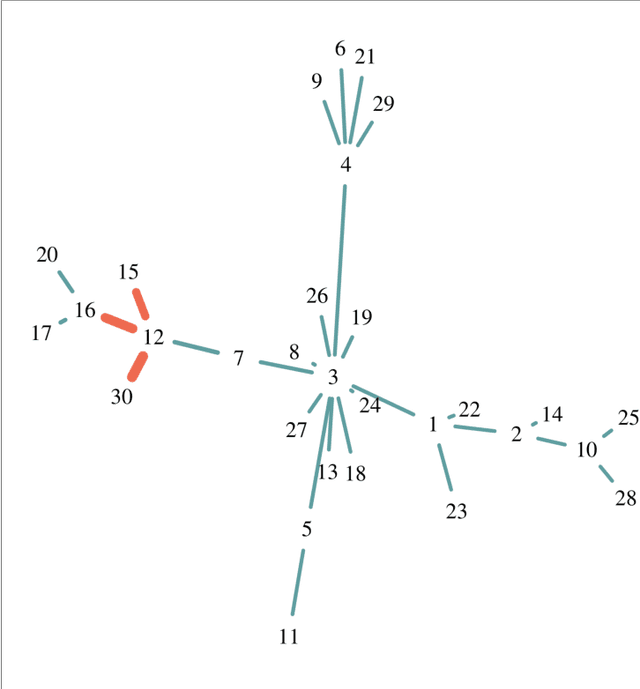
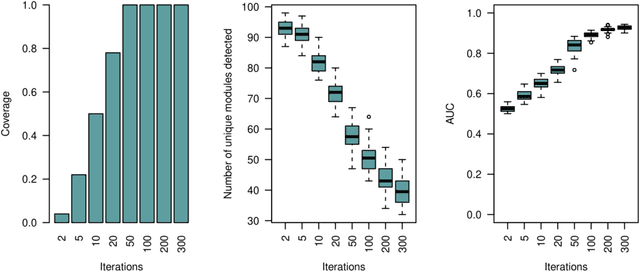
Network-based algorithms are used in most domains of research and industry in a wide variety of applications and are of great practical use. In this work, we demonstrate subnetwork detection based on multi-modal node features using a new Greedy Decision Forest for better interpretability. The latter will be a crucial factor in retaining experts and gaining their trust in such algorithms in the future. To demonstrate a concrete application example, we focus in this paper on bioinformatics and systems biology with a special focus on biomedicine. However, our methodological approach is applicable in many other domains as well. Systems biology serves as a very good example of a field in which statistical data-driven machine learning enables the analysis of large amounts of multi-modal biomedical data. This is important to reach the future goal of precision medicine, where the complexity of patients is modeled on a system level to best tailor medical decisions, health practices and therapies to the individual patient. Our glass-box approach could help to uncover disease-causing network modules from multi-omics data to better understand diseases such as cancer.