 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEVA: Towards a universal model of the immune system
Feb 12, 2026The effective application of foundation models to translational research in immune-mediated diseases requires multimodal patient-level representations that can capture complex phenotypes emerging from multicellular interactions. Yet most current biological foundation models focus only on single-cell resolution and are evaluated on technical metrics often disconnected from actual drug development tasks and challenges. Here, we introduce EVA, the first cross-species, multimodal foundation model of immunology and inflammation, a therapeutic area where shared pathogenic mechanisms create unique opportunities for transfer learning. EVA harmonizes transcriptomics data across species, platforms, and resolutions, and integrates histology data to produce rich, unified patient representations. We establish clear scaling laws, demonstrating that increasing model size and compute translates to improvements in both pretraining and downstream tasks performance. We introduce a comprehensive evaluation suite of 39 tasks spanning the drug development pipeline: zero-shot target efficacy and gene function prediction for discovery, cross-species or cross-diseases molecular perturbations for preclinical development, and patient stratification with treatment response prediction or disease activity prediction for clinical trials applications. We benchmark EVA against several state-of-the-art biological foundation models and baselines on these tasks, and demonstrate state-of-the-art results on each task category. Using mechanistic interpretability, we further identify biological meaningful features, revealing intertwined representations across species and technologies. We release an open version of EVA for transcriptomics to accelerate research on immune-mediated diseases.
Domain Adaptation Principal Component Analysis: base linear method for learning with out-of-distribution data
Aug 28, 2022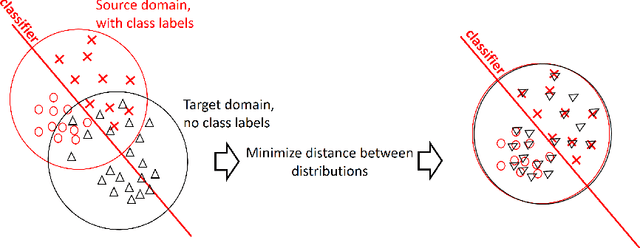
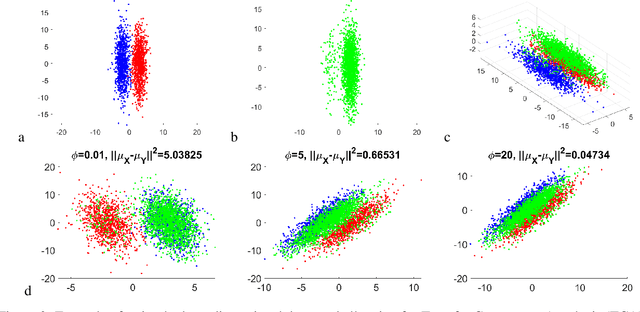
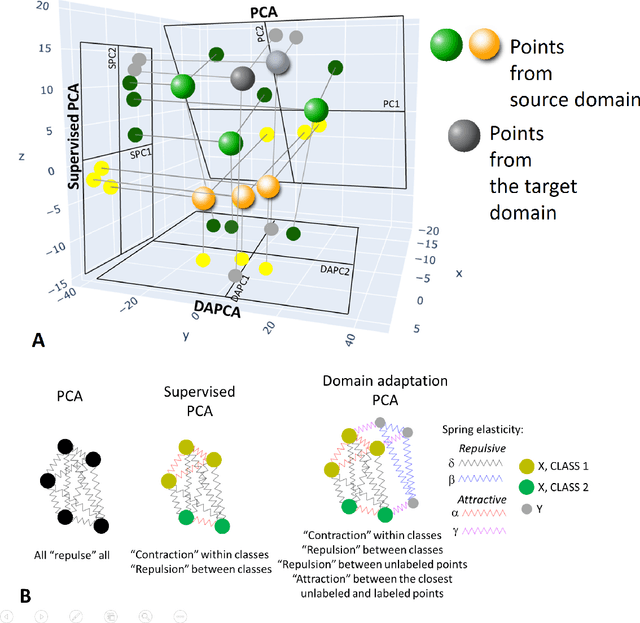
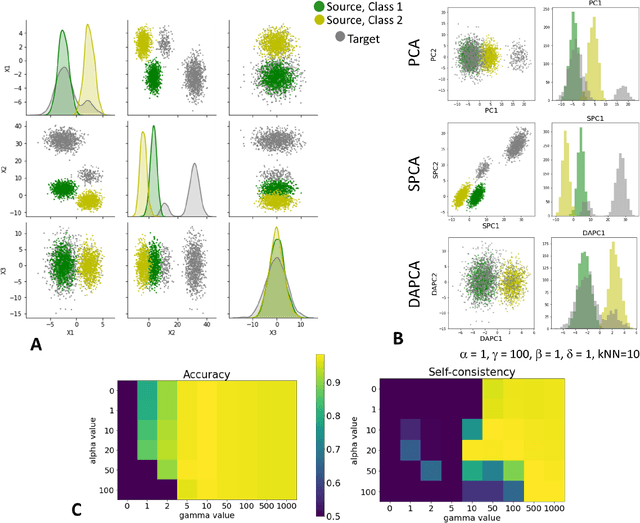
Domain adaptation is a popular paradigm in modern machine learning which aims at tackling the problem of divergence between training or validation dataset possessing labels for learning and testing a classifier (source domain) and a potentially large unlabeled dataset where the model is exploited (target domain). The task is to find such a common representation of both source and target datasets in which the source dataset is informative for training and such that the divergence between source and target would be minimized. Most popular solutions for domain adaptation are currently based on training neural networks that combine classification and adversarial learning modules, which are data hungry and usually difficult to train. We present a method called Domain Adaptation Principal Component Analysis (DAPCA) which finds a linear reduced data representation useful for solving the domain adaptation task. DAPCA is based on introducing positive and negative weights between pairs of data points and generalizes the supervised extension of principal component analysis. DAPCA represents an iterative algorithm such that at each iteration a simple quadratic optimization problem is solved. The convergence of the algorithm is guaranteed and the number of iterations is small in practice. We validate the suggested algorithm on previously proposed benchmarks for solving the domain adaptation task, and also show the benefit of using DAPCA in the analysis of single cell omics datasets in biomedical applications. Overall, DAPCA can serve as a useful preprocessing step in many machine learning applications leading to reduced dataset representations, taking into account possible divergence between source and target domains.