 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrediction of Atomization Energy Using Graph Kernel and Active Learning
Paper and Code
Oct 18, 2018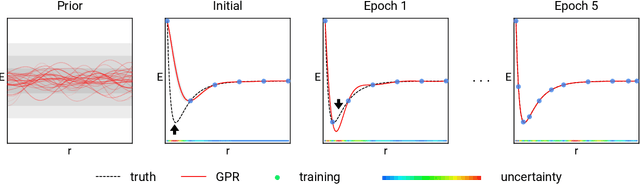
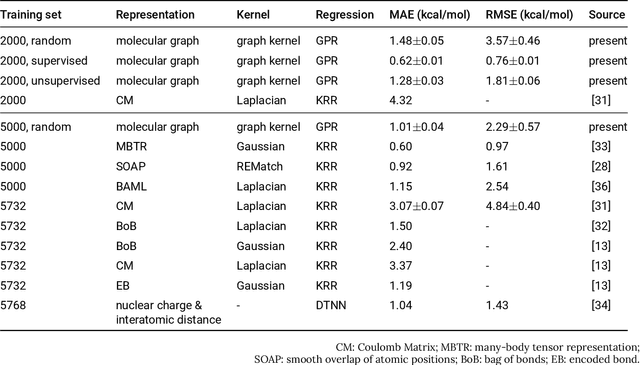
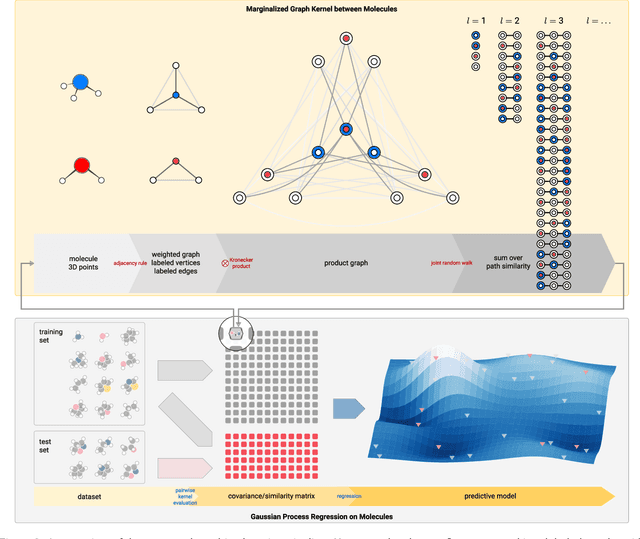
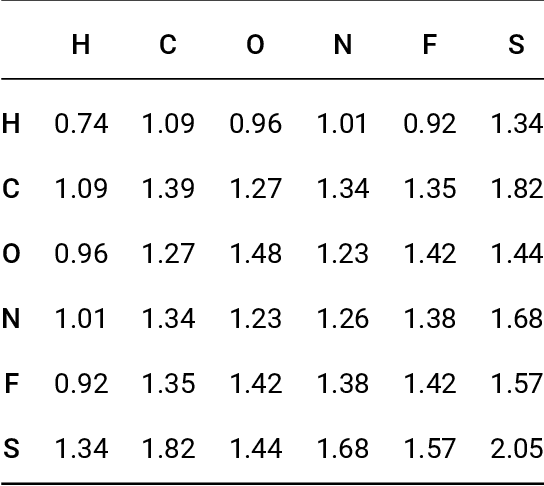
Data-driven prediction of molecular properties presents unique challenges to the design of machine learning methods concerning data structure/dimensionality, symmetry adaption, and confidence management. In this paper, we present a kernel-based pipeline that can learn and predict the atomization energy of molecules with high accuracy. The framework employs Gaussian process regression to perform predictions based on the similarity between molecules, which is computed using the marginalized graph kernel. We discuss why the graph kernel, paired with a graph representation of the molecules, is particularly useful for predicting extensive properties. We demonstrate that using an active learning procedure, the proposed method can achieve a mean absolute error less than 1.0 kcal/mol on the QM7 data set using as few as 1200 training samples and 1 hour of training time. This is a demonstration, in contrast to common believes, that regression models based on kernel methods can be simultaneously accurate and fast predictors.