 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInferring a Continuous Distribution of Atom Coordinates from Cryo-EM Images using VAEs
Paper and Code
Jun 26, 2021
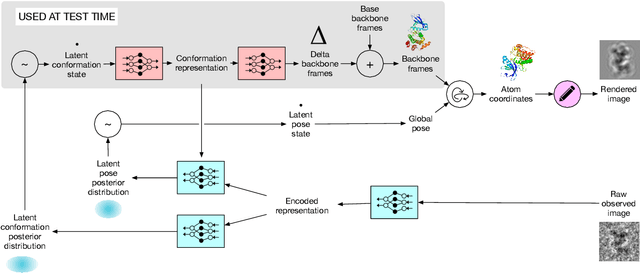
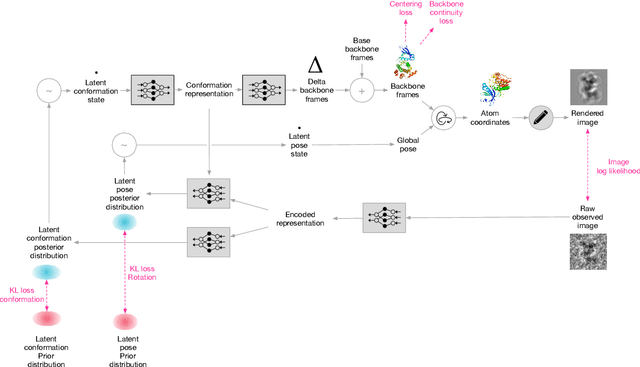
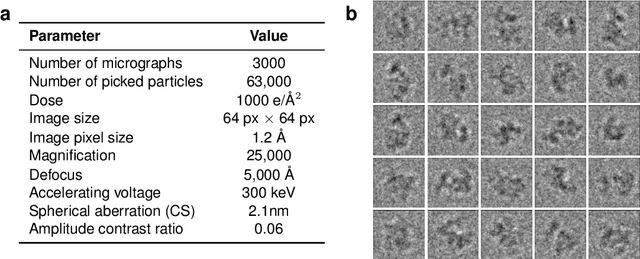
Cryo-electron microscopy (cryo-EM) has revolutionized experimental protein structure determination. Despite advances in high resolution reconstruction, a majority of cryo-EM experiments provide either a single state of the studied macromolecule, or a relatively small number of its conformations. This reduces the effectiveness of the technique for proteins with flexible regions, which are known to play a key role in protein function. Recent methods for capturing conformational heterogeneity in cryo-EM data model it in volume space, making recovery of continuous atomic structures challenging. Here we present a fully deep-learning-based approach using variational auto-encoders (VAEs) to recover a continuous distribution of atomic protein structures and poses directly from picked particle images and demonstrate its efficacy on realistic simulated data. We hope that methods built on this work will allow incorporation of stronger prior information about protein structure and enable better understanding of non-rigid protein structures.