 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Network for Hamiltonian-Based Material Property Prediction
Paper and Code
May 27, 2020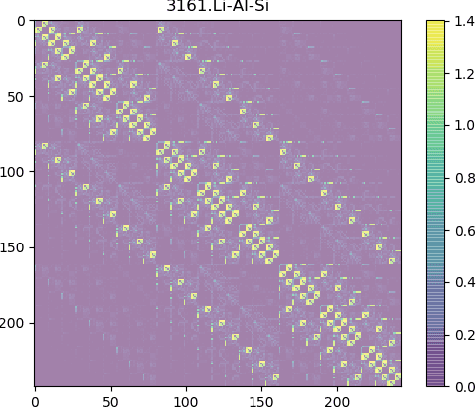

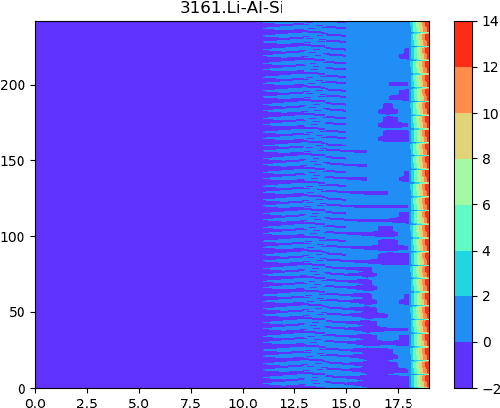
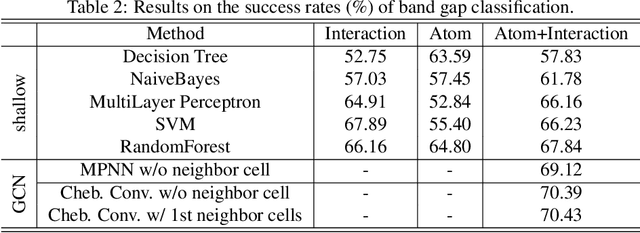
Development of next-generation electronic devices for applications call for the discovery of quantum materials hosting novel electronic, magnetic, and topological properties. Traditional electronic structure methods require expensive computation time and memory consumption, thus a fast and accurate prediction model is desired with increasing importance. Representing the interactions among atomic orbitals in any material, a material Hamiltonian provides all the essential elements that control the structure-property correlations in inorganic compounds. Effective learning of material Hamiltonian by developing machine learning methodologies therefore offers a transformative approach to accelerate the discovery and design of quantum materials. With this motivation, we present and compare several different graph convolution networks that are able to predict the band gap for inorganic materials. The models are developed to incorporate two different features: the information of each orbital itself and the interaction between each other. The information of each orbital includes the name, relative coordinates with respect to the center of super cell and the atom number, while the interaction between orbitals are represented by the Hamiltonian matrix. The results show that our model can get a promising prediction accuracy with cross-validation.