 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstanding When Poisson Log-Normal Models Outperform Penalized Poisson Regression for Microbiome Count Data
Apr 04, 2026Multivariate count models are often justified by their ability to capture latent dependence, but researchers receive little guidance on when this added structure improves on simpler penalized marginal Poisson regression. We study this question using real microbiome data under a unified held-out evaluation framework. For count prediction, we compare PLN and GLMNet(Poisson) on 20 datasets spanning 32 to 18,270 samples and 24 to 257 taxa, using held-out Poisson deviance under leave-one-taxon-out prediction with 3-fold sample cross-validation rather than synthetic or in-sample criteria. For network inference, we compare PLNNetwork and GLMNet(Poisson) neighborhood selection on five publicly available datasets with experimentally validated microbial interaction truth. PLN outperforms GLMNet(Poisson) on most count-prediction datasets, with gains up to 38 percent. The primary predictor of the winner is the sample-to-taxon ratio, with mean absolute correlation as the strongest secondary signal and overdispersion as an additional predictor. PLNNetwork performs best on broad undirected interaction benchmarks, whereas GLMNet(Poisson) is better aligned with local or directional effects. Taken together, these results provide guidance for choosing between latent multivariate count models and penalized Poisson regression in biological count prediction and interaction recovery.
Interval Regression: A Comparative Study with Proposed Models
Mar 03, 2025



Regression models are essential for a wide range of real-world applications. However, in practice, target values are not always precisely known; instead, they may be represented as intervals of acceptable values. This challenge has led to the development of Interval Regression models. In this study, we provide a comprehensive review of existing Interval Regression models and introduce alternative models for comparative analysis. Experiments are conducted on both real-world and synthetic datasets to offer a broad perspective on model performance. The results demonstrate that no single model is universally optimal, highlighting the importance of selecting the most suitable model for each specific scenario.
Efficient line search for optimizing Area Under the ROC Curve in gradient descent
Oct 11, 2024



Receiver Operating Characteristic (ROC) curves are useful for evaluation in binary classification and changepoint detection, but difficult to use for learning since the Area Under the Curve (AUC) is piecewise constant (gradient zero almost everywhere). Recently the Area Under Min (AUM) of false positive and false negative rates has been proposed as a differentiable surrogate for AUC. In this paper we study the piecewise linear/constant nature of the AUM/AUC, and propose new efficient path-following algorithms for choosing the learning rate which is optimal for each step of gradient descent (line search), when optimizing a linear model. Remarkably, our proposed line search algorithm has the same log-linear asymptotic time complexity as gradient descent with constant step size, but it computes a complete representation of the AUM/AUC as a function of step size. In our empirical study of binary classification problems, we verify that our proposed algorithm is fast and exact; in changepoint detection problems we show that the proposed algorithm is just as accurate as grid search, but faster.
SOAK: Same/Other/All K-fold cross-validation for estimating similarity of patterns in data subsets
Oct 11, 2024



In many real-world applications of machine learning, we are interested to know if it is possible to train on the data that we have gathered so far, and obtain accurate predictions on a new test data subset that is qualitatively different in some respect (time period, geographic region, etc). Another question is whether data subsets are similar enough so that it is beneficial to combine subsets during model training. We propose SOAK, Same/Other/All K-fold cross-validation, a new method which can be used to answer both questions. SOAK systematically compares models which are trained on different subsets of data, and then used for prediction on a fixed test subset, to estimate the similarity of learnable/predictable patterns in data subsets. We show results of using SOAK on six new real data sets (with geographic/temporal subsets, to check if predictions are accurate on new subsets), 3 image pair data sets (subsets are different image types, to check that we get smaller prediction error on similar images), and 11 benchmark data sets with predefined train/test splits (to check similarity of predefined splits).
Finite Sample Complexity Analysis of Binary Segmentation
Oct 11, 2024Binary segmentation is the classic greedy algorithm which recursively splits a sequential data set by optimizing some loss or likelihood function. Binary segmentation is widely used for changepoint detection in data sets measured over space or time, and as a sub-routine for decision tree learning. In theory it should be extremely fast for $N$ data and $K$ splits, $O(N K)$ in the worst case, and $O(N \log K)$ in the best case. In this paper we describe new methods for analyzing the time and space complexity of binary segmentation for a given finite $N$, $K$, and minimum segment length parameter. First, we describe algorithms that can be used to compute the best and worst case number of splits the algorithm must consider. Second, we describe synthetic data that achieve the best and worst case and which can be used to test for correct implementation of the algorithm. Finally, we provide an empirical analysis of real data which suggests that binary segmentation is often close to optimal speed in practice.
Deep Learning Approach for Changepoint Detection: Penalty Parameter Optimization
Aug 01, 2024Changepoint detection, a technique for identifying significant shifts within data sequences, is crucial in various fields such as finance, genomics, medicine, etc. Dynamic programming changepoint detection algorithms are employed to identify the locations of changepoints within a sequence, which rely on a penalty parameter to regulate the number of changepoints. To estimate this penalty parameter, previous work uses simple models such as linear models or decision trees. This study introduces a novel deep learning method for predicting penalty parameters, leading to demonstrably improved changepoint detection accuracy on large benchmark supervised labeled datasets compared to previous methods.
Cross-Validation for Training and Testing Co-occurrence Network Inference Algorithms
Sep 26, 2023Microorganisms are found in almost every environment, including the soil, water, air, and inside other organisms, like animals and plants. While some microorganisms cause diseases, most of them help in biological processes such as decomposition, fermentation and nutrient cycling. A lot of research has gone into studying microbial communities in various environments and how their interactions and relationships can provide insights into various diseases. Co-occurrence network inference algorithms help us understand the complex associations of micro-organisms, especially bacteria. Existing network inference algorithms employ techniques such as correlation, regularized linear regression, and conditional dependence, which have different hyper-parameters that determine the sparsity of the network. Previous methods for evaluating the quality of the inferred network include using external data, and network consistency across sub-samples, both which have several drawbacks that limit their applicability in real microbiome composition data sets. We propose a novel cross-validation method to evaluate co-occurrence network inference algorithms, and new methods for applying existing algorithms to predict on test data. Our empirical study shows that the proposed method is useful for hyper-parameter selection (training) and comparing the quality of the inferred networks between different algorithms (testing).
Optimizing ROC Curves with a Sort-Based Surrogate Loss Function for Binary Classification and Changepoint Detection
Jul 02, 2021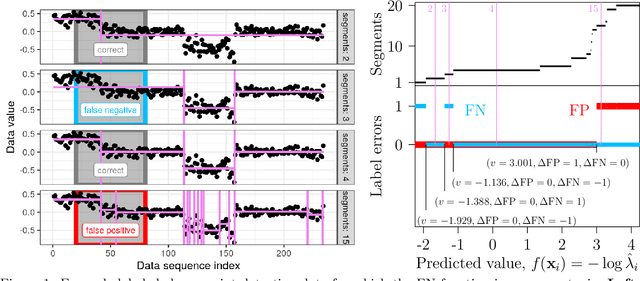
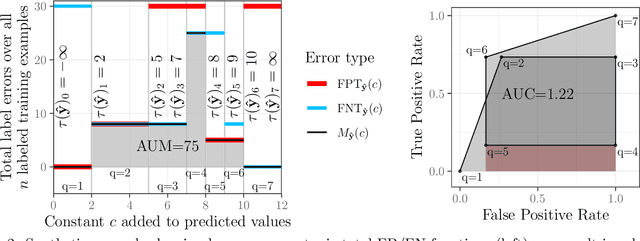
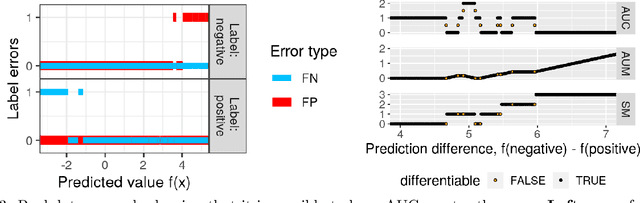
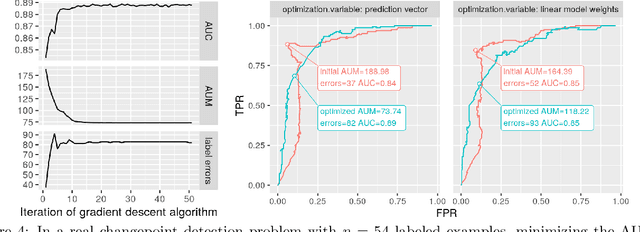
Receiver Operating Characteristic (ROC) curves are plots of true positive rate versus false positive rate which are useful for evaluating binary classification models, but difficult to use for learning since the Area Under the Curve (AUC) is non-convex. ROC curves can also be used in other problems that have false positive and true positive rates such as changepoint detection. We show that in this more general context, the ROC curve can have loops, points with highly sub-optimal error rates, and AUC greater than one. This observation motivates a new optimization objective: rather than maximizing the AUC, we would like a monotonic ROC curve with AUC=1 that avoids points with large values for Min(FP,FN). We propose a convex relaxation of this objective that results in a new surrogate loss function called the AUM, short for Area Under Min(FP, FN). Whereas previous loss functions are based on summing over all labeled examples or pairs, the AUM requires a sort and a sum over the sequence of points on the ROC curve. We show that AUM directional derivatives can be efficiently computed and used in a gradient descent learning algorithm. In our empirical study of supervised binary classification and changepoint detection problems, we show that our new AUM minimization learning algorithm results in improved AUC and comparable speed relative to previous baselines.
Increased peak detection accuracy in over-dispersed ChIP-seq data with supervised segmentation models
Dec 15, 2020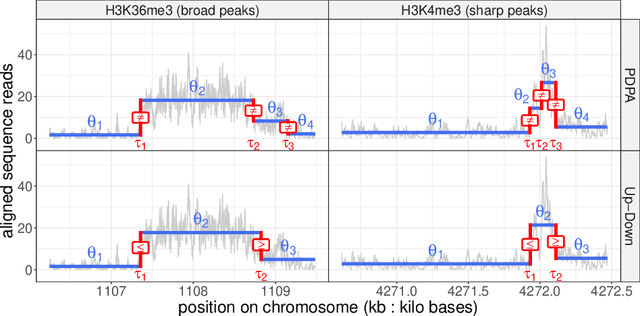
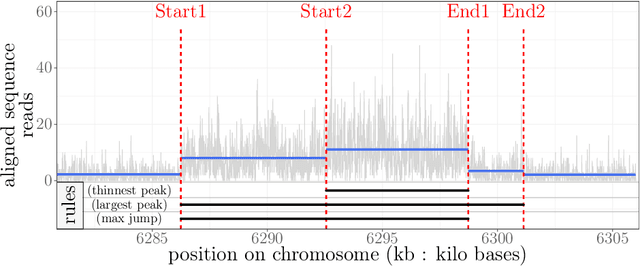
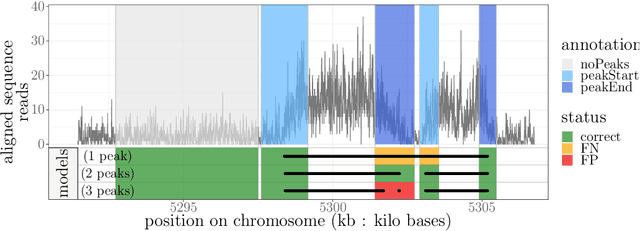
Motivation: Histone modification constitutes a basic mechanism for the genetic regulation of gene expression. In early 2000s, a powerful technique has emerged that couples chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq). This technique provides a direct survey of the DNA regions associated to these modifications. In order to realize the full potential of this technique, increasingly sophisticated statistical algorithms have been developed or adapted to analyze the massive amount of data it generates. Many of these algorithms were built around natural assumptions such as the Poisson one to model the noise in the count data. In this work we start from these natural assumptions and show that it is possible to improve upon them. Results: The results of our comparisons on seven reference datasets of histone modifications (H3K36me3 and H3K4me3) suggest that natural assumptions are not always realistic under application conditions. We show that the unconstrained multiple changepoint detection model, with alternative noise assumptions and a suitable setup, reduces the over-dispersion exhibited by count data and turns out to detect peaks more accurately than algorithms which rely on these natural assumptions.
Labeled Optimal Partitioning
Jun 24, 2020
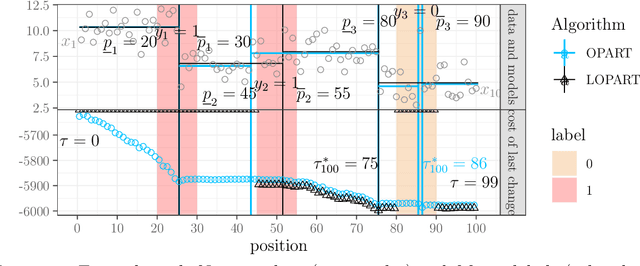
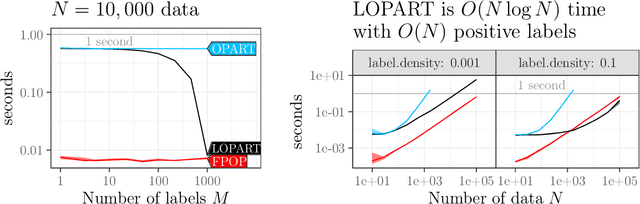
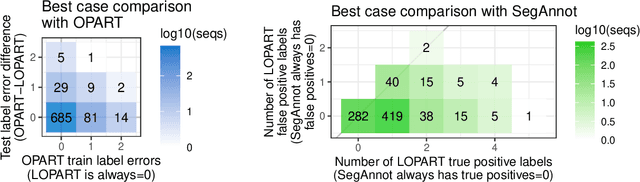
In data sequences measured over space or time, an important problem is accurate detection of abrupt changes. In partially labeled data, it is important to correctly predict presence/absence of changes in positive/negative labeled regions, in both the train and test sets. One existing dynamic programming algorithm is designed for prediction in unlabeled test regions (and ignores the labels in the train set); another is for accurate fitting of train labels (but does not predict changepoints in unlabeled test regions). We resolve these issues by proposing a new optimal changepoint detection model that is guaranteed to fit the labels in the train data, and can also provide predictions of unlabeled changepoints in test data. We propose a new dynamic programming algorithm, Labeled Optimal Partitioning (LOPART), and we provide a formal proof that it solves the resulting non-convex optimization problem. We provide theoretical and empirical analysis of the time complexity of our algorithm, in terms of the number of labels and the size of the data sequence to segment. Finally, we provide empirical evidence that our algorithm is more accurate than the existing baselines, in terms of train and test label error.