 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI-Driven Expansion and Application of the Alexandria Database
Dec 09, 2025



We present a novel multi-stage workflow for computational materials discovery that achieves a 99% success rate in identifying compounds within 100 meV/atom of thermodynamic stability, with a threefold improvement over previous approaches. By combining the Matra-Genoa generative model, Orb-v2 universal machine learning interatomic potential, and ALIGNN graph neural network for energy prediction, we generated 119 million candidate structures and added 1.3 million DFT-validated compounds to the ALEXANDRIA database, including 74 thousand new stable materials. The expanded ALEXANDRIA database now contains 5.8 million structures with 175 thousand compounds on the convex hull. Predicted structural disorder rates (37-43%) match experimental databases, unlike other recent AI-generated datasets. Analysis reveals fundamental patterns in space group distributions, coordination environments, and phase stability networks, including sub-linear scaling of convex hull connectivity. We release the complete dataset, including sAlex25 with 14 million out-of-equilibrium structures containing forces and stresses for training universal force fields. We demonstrate that fine-tuning a GRACE model on this data improves benchmark accuracy. All data, models, and workflows are freely available under Creative Commons licenses.
Large-scale machine-learning-assisted exploration of the whole materials space
Oct 02, 2022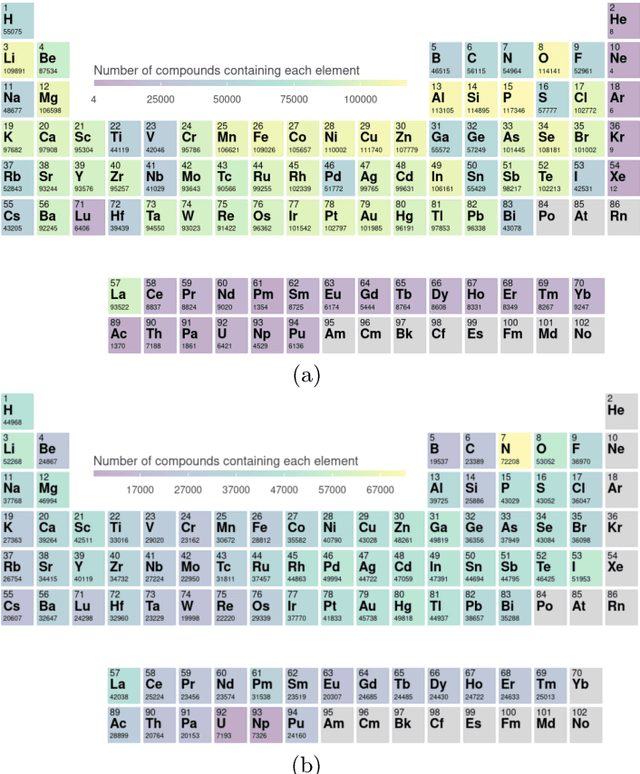
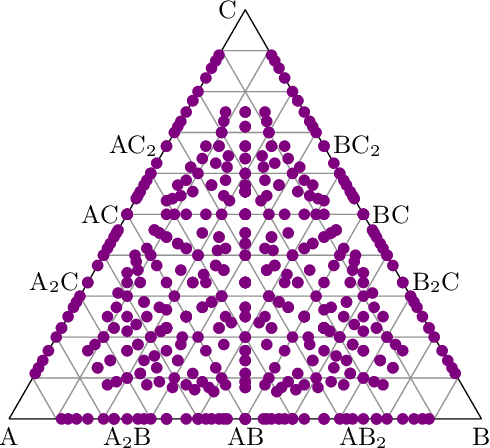
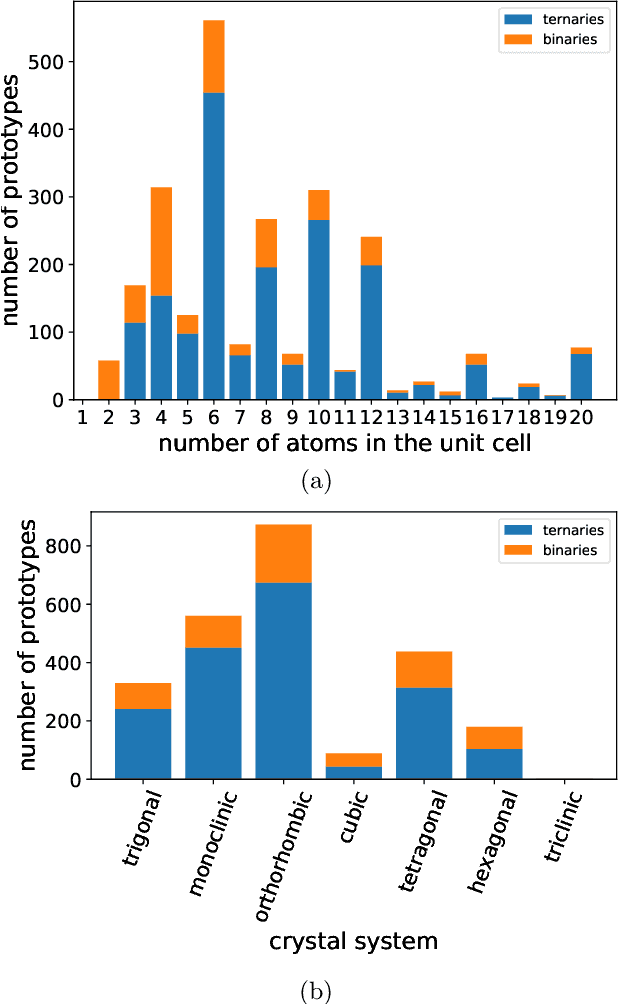
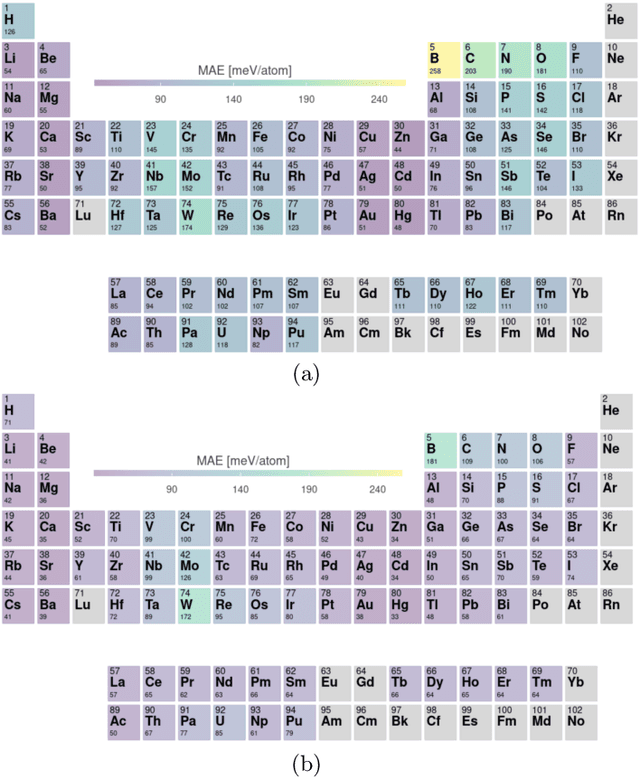
Crystal-graph attention networks have emerged recently as remarkable tools for the prediction of thermodynamic stability and materials properties from unrelaxed crystal structures. Previous networks trained on two million materials exhibited, however, strong biases originating from underrepresented chemical elements and structural prototypes in the available data. We tackled this issue computing additional data to provide better balance across both chemical and crystal-symmetry space. Crystal-graph networks trained with this new data show unprecedented generalization accuracy, and allow for reliable, accelerated exploration of the whole space of inorganic compounds. We applied this universal network to perform machine-learning assisted high-throughput materials searches including 2500 binary and ternary structure prototypes and spanning about 1 billion compounds. After validation using density-functional theory, we uncover in total 19512 additional materials on the convex hull of thermodynamic stability and ~150000 compounds with a distance of less than 50 meV/atom from the hull. Combining again machine learning and ab-initio methods, we finally evaluate the discovered materials for applications as superconductors, superhard materials, and we look for candidates with large gap deformation potentials, finding several compounds with extreme values of these properties.