 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA multitask transfer learning framework for the prediction of virus-human protein-protein interactions
Nov 26, 2021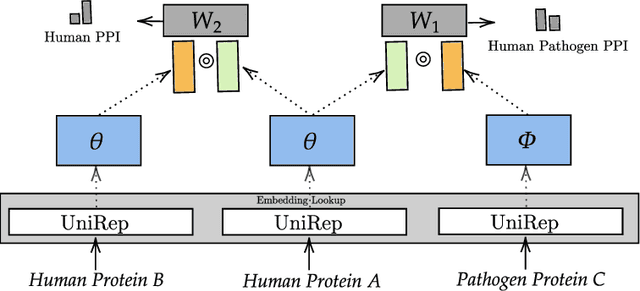
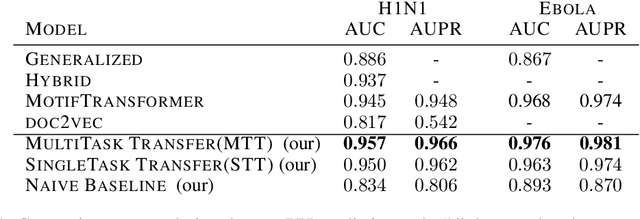
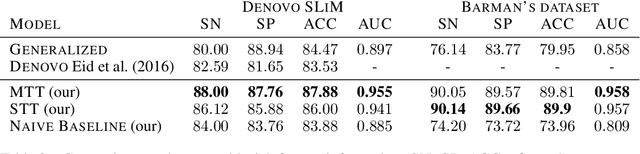

Viral infections are causing significant morbidity and mortality worldwide. Understanding the interaction patterns between a particular virus and human proteins plays a crucial role in unveiling the underlying mechanism of viral infection and pathogenesis. This could further help in the prevention and treatment of virus-related diseases. However, the task of predicting protein-protein interactions between a new virus and human cells is extremely challenging due to scarce data on virus-human interactions and fast mutation rates of most viruses. We developed a multitask transfer learning approach that exploits the information of around 24 million protein sequences and the interaction patterns from the human interactome to counter the problem of small training datasets. Instead of using hand-crafted protein features, we utilize statistically rich protein representations learned by a deep language modeling approach from a massive source of protein sequences. Additionally, we employ an additional objective which aims to maximize the probability of observing human protein-protein interactions. This additional task objective acts as a regularizer and also allows to incorporate domain knowledge to inform the virus-human protein-protein interaction prediction model. Our approach achieved competitive results on 13 benchmark datasets and the case study for the SAR-CoV-2 virus receptor. Experimental results show that our proposed model works effectively for both virus-human and bacteria-human protein-protein interaction prediction tasks. We share our code for reproducibility and future research at https://git.l3s.uni-hannover.de/dong/multitask-transfer.
MuCoMiD: A Multitask Convolutional Learning Framework for miRNA-Disease Association Prediction
Aug 08, 2021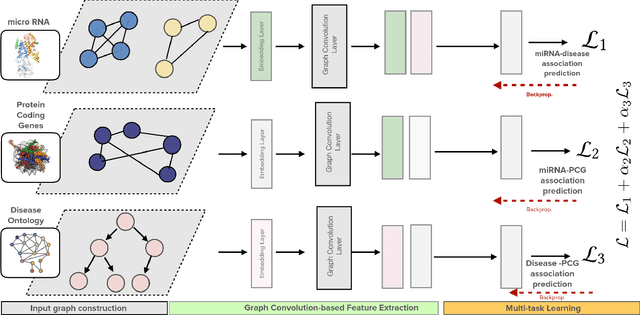


Growing evidence from recent studies implies that microRNA or miRNA could serve as biomarkers in various complex human diseases. Since wet-lab experiments are expensive and time-consuming, computational techniques for miRNA-disease association prediction have attracted a lot of attention in recent years. Data scarcity is one of the major challenges in building reliable machine learning models. Data scarcity combined with the use of pre-calculated hand-crafted input features has led to problems of overfitting and data leakage. We overcome the limitations of existing works by proposing a novel multi-tasking convolution-based approach, which we refer to as MuCoMiD. MuCoMiD allows automatic feature extraction while incorporating knowledge from 4 heterogeneous biological information sources (interactions between miRNA/diseases and protein-coding genes (PCG), miRNA family information, and disease ontology) in a multi-task setting which is a novel perspective and has not been studied before. The use of multi-channel convolutions allows us to extract expressive representations while keeping the model linear and, therefore, simple. To effectively test the generalization capability of our model, we construct large-scale experiments on standard benchmark datasets as well as our proposed larger independent test sets and case studies. MuCoMiD shows an improvement of at least 5% in 5-fold CV evaluation on HMDDv2.0 and HMDDv3.0 datasets and at least 49% on larger independent test sets with unseen miRNA and diseases over state-of-the-art approaches. We share our code for reproducibility and future research at https://git.l3s.uni-hannover.de/dong/cmtt.