 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTheodore Perkins
Is graph biased feature selection of genes better than random?
Oct 21, 2019
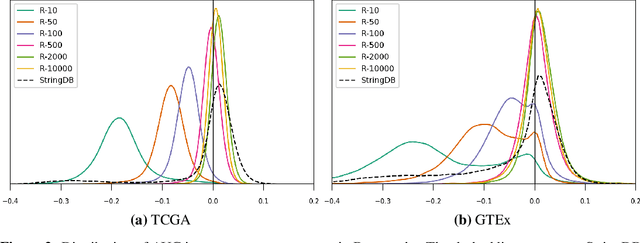
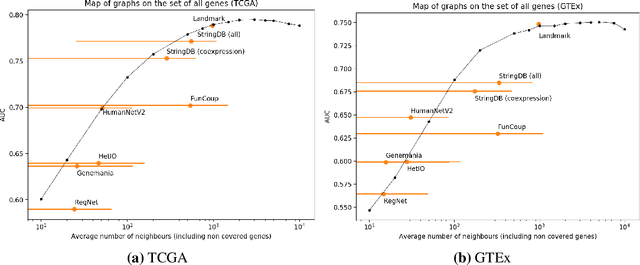
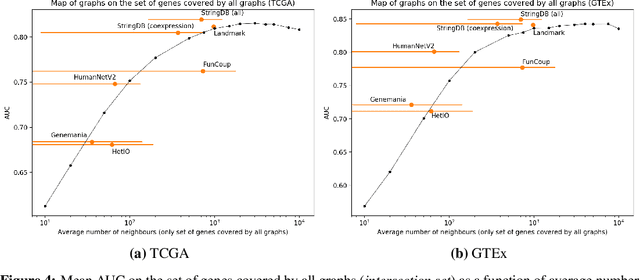
Gene interaction graphs aim to capture various relationships between genes and can represent decades of biology research. When trying to make predictions from genomic data, those graphs could be used to overcome the curse of dimensionality by making machine learning models sparser and more biased with biological common knowledge. In this work, we focus on assessing whether those graphs capture dependencies seen in gene expression data better than random. We formulate a condition that graphs should satisfy to provide a good bias and propose to test it using a 'Single Gene Inference' (SGI) task. We compare random graphs with seven major gene interaction graphs published by different research groups, aiming to measure the true benefit of using biologically relevant graphs in this context. Our analysis finds that dependencies can be captured almost as well at random which suggests that, in terms of gene expression levels, the relevant information about the state of the cell is spread across many genes.