 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Syntax to Semantics: Unveiling the Emergence of Chirality in SMILES Translation Models
May 11, 2026Understanding how chemical language models (CLMs) learn chemical meaning from molecular string representations, rather than only surface-level string patterns, is an important question in chemical representation learning and machine learning for chemistry. Chirality provides a demanding test case: enantiomers can differ greatly in pharmacological activity and toxicity, yet CLMs often struggle to distinguish chiral configurations reliably. Here we present Pan-CORE (Pan-Chemical Omniscale Representation Engine), a family of autoregressive Transformer-based encoder-decoder models for SMILES translation, and use high-temporal-resolution checkpoint analysis to investigate how chiral information is learned during training. Across all tested Pan-CORE variants, we observe a reproducible jump-up in which chiral-token accuracy rises abruptly after a long plateau, suggesting that chiral learning stagnation is not explained by model capacity alone and instead reflects the complexity of chiral constraints. Analyses of attention dynamics, residual-stream trajectories, and latent-space geometry support an encoder-centered mechanism in which chiral-token representations undergo transient destabilization and reconstruction, seen as a V-shaped drop and recovery in vector norm and directional stability, together with a clear reorganization of chiral molecular representations in the latent space. Encoder-decoder cross-evaluation further supports the encoder-centered nature of the transition, and targeted attention-head ablation identifies a small set of chiral-sensitive heads whose removal selectively reduces chiral-token accuracy even in the fully trained model. These findings show that SMILES translation can serve as a useful experimental system for mechanistic analysis of semantic emergence in CLMs, with implications for interpretable chemical representation learning.
A novel molecule generative model of VAE combined with Transformer
Feb 19, 2024Recently, molecule generation using deep learning has been actively investigated in drug discovery. In this field, Transformer and VAE are widely used as powerful models, but they are rarely used in combination due to structural and performance mismatch of them. This study proposes a model that combines these two models through structural and parameter optimization in handling diverse molecules. The proposed model shows comparable performance to existing models in generating molecules, and showed by far superior performance in generating molecules with unseen structures. In addition, the proposed model successfully predicted molecular properties using the latent representation of VAE. Ablation studies suggested the advantage of VAE over other generative models like language model in generating novel molecules, and that the molecules can be described by ~32 dimensional variables, much smaller than existing descriptors and models. This study is expected to provide a virtual chemical library containing a wide variety of compounds for virtual screening and to enable efficient screening.
Difficulty in learning chirality for Transformer fed with SMILES
Mar 21, 2023Recent years have seen development of descriptor generation based on representation learning of extremely diverse molecules, especially those that apply natural language processing (NLP) models to SMILES, a literal representation of molecular structure. However, little research has been done on how these models understand chemical structure. To address this, we investigated the relationship between the learning progress of SMILES and chemical structure using a representative NLP model, the Transformer. The results suggest that while the Transformer learns partial structures of molecules quickly, it requires extended training to understand overall structures. Consistently, the accuracy of molecular property predictions using descriptors generated from models at different learning steps was similar from the beginning to the end of training. Furthermore, we found that the Transformer requires particularly long training to learn chirality and sometimes stagnates with low translation accuracy due to misunderstanding of enantiomers. These findings are expected to deepen understanding of NLP models in chemistry.
Investigation of chemical structure recognition by encoder-decoder models in learning progress
Nov 02, 2022Descriptor generation methods using latent representations of encoder$-$decoder (ED) models with SMILES as input are useful because of the continuity of descriptor and restorability to the structure. However, it is not clear how the structure is recognized in the learning progress of ED models. In this work, we created ED models of various learning progress and investigated the relationship between structural information and learning progress. We showed that compound substructures were learned early in ED models by monitoring the accuracy of downstream tasks and input$-$output substructure similarity using substructure$-$based descriptors, which suggests that existing evaluation methods based on the accuracy of downstream tasks may not be sensitive enough to evaluate the performance of ED models with SMILES as descriptor generation methods. On the other hand, we showed that structure restoration was time$-$consuming, and in particular, insufficient learning led to the estimation of a larger structure than the actual one. It can be inferred that determining the endpoint of the structure is a difficult task for the model. To our knowledge, this is the first study to link the learning progress of SMILES by ED model to chemical structures for a wide range of chemicals.
NRBdMF: A recommendation algorithm for predicting drug effects considering directionality
Aug 05, 2022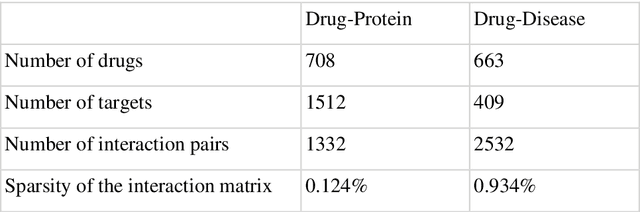
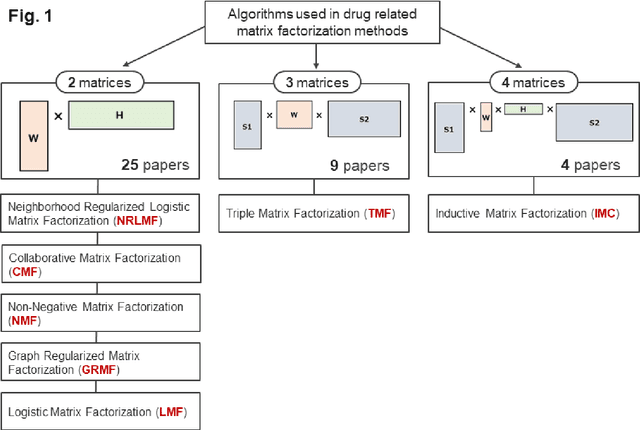
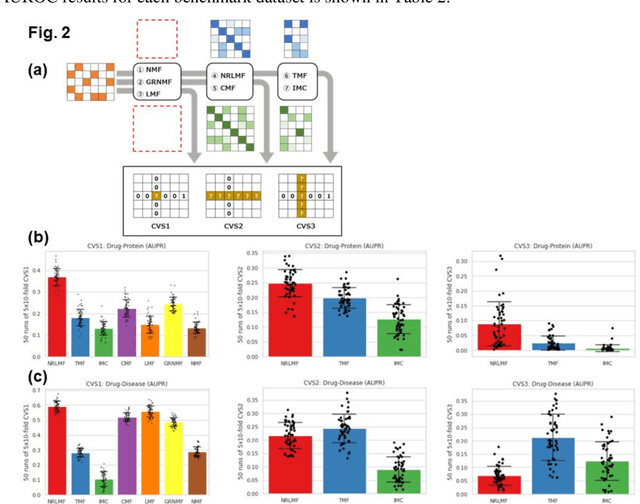
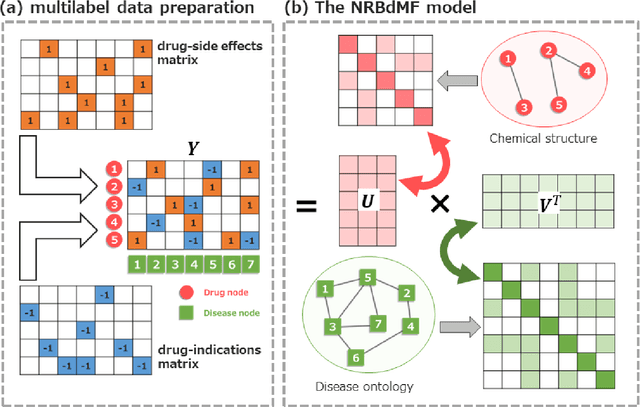
Predicting the novel effects of drugs based on information about approved drugs can be regarded as a recommendation system. Matrix factorization is one of the most used recommendation systems and various algorithms have been devised for it. A literature survey and summary of existing algorithms for predicting drug effects demonstrated that most such methods, including neighborhood regularized logistic matrix factorization, which was the best performer in benchmark tests, used a binary matrix that considers only the presence or absence of interactions. However, drug effects are known to have two opposite aspects, such as side effects and therapeutic effects. In the present study, we proposed using neighborhood regularized bidirectional matrix factorization (NRBdMF) to predict drug effects by incorporating bidirectionality, which is a characteristic property of drug effects. We used this proposed method for predicting side effects using a matrix that considered the bidirectionality of drug effects, in which known side effects were assigned a positive label (plus 1) and known treatment effects were assigned a negative (minus 1) label. The NRBdMF model, which utilizes drug bidirectional information, achieved enrichment of side effects at the top and indications at the bottom of the prediction list. This first attempt to consider the bidirectional nature of drug effects using NRBdMF showed that it reduced false positives and produced a highly interpretable output.
Investigation of a Data Split Strategy Involving the Time Axis in Adverse Event Prediction Using Machine Learning
Apr 19, 2022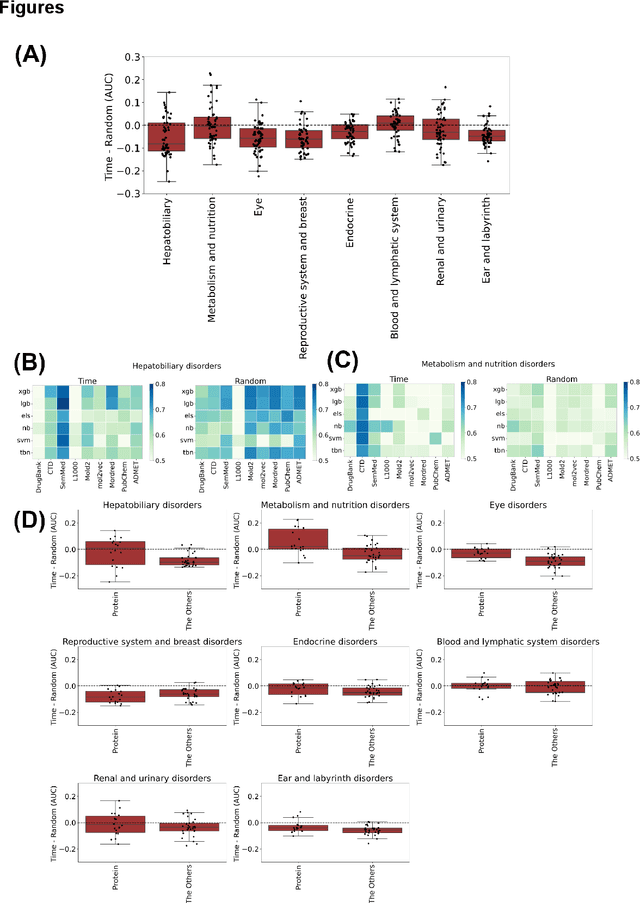
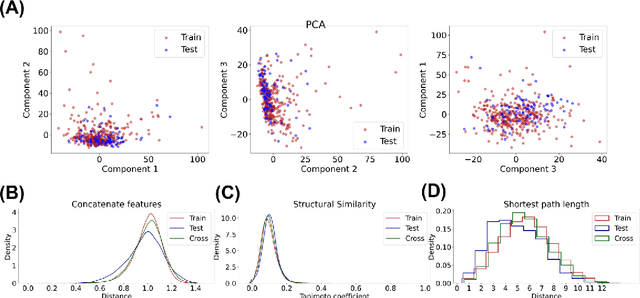
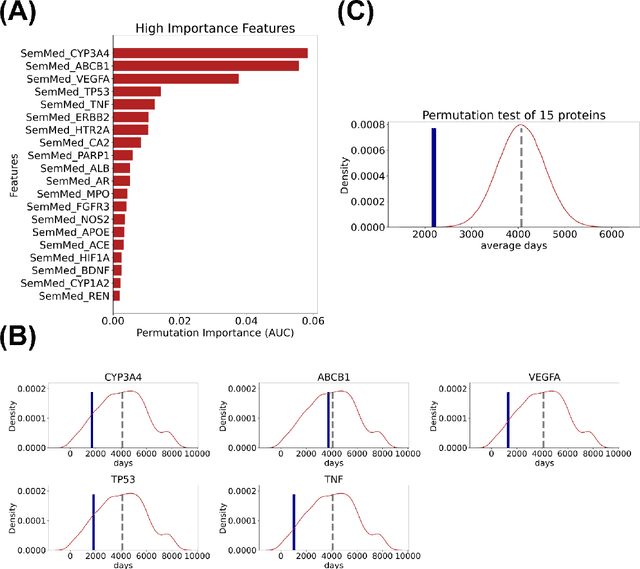
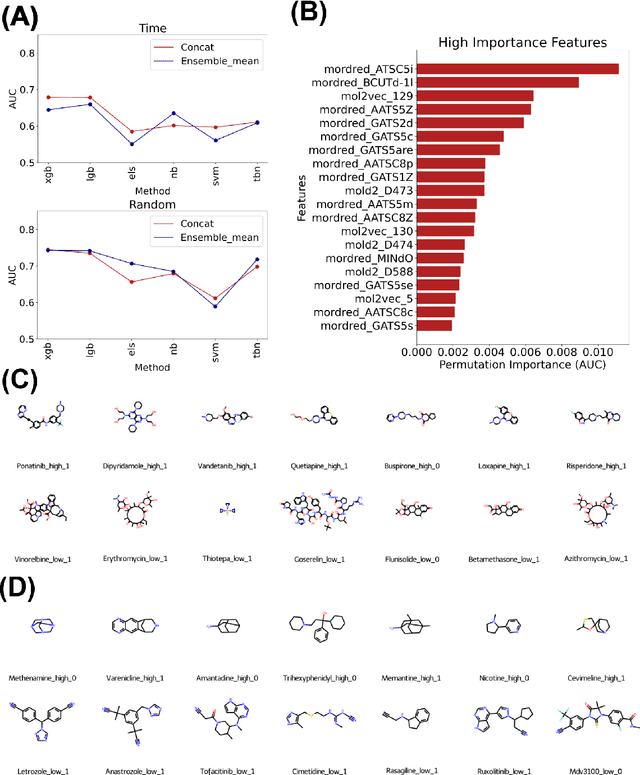
Adverse events are a serious issue in drug development and many prediction methods using machine learning have been developed. The random split cross-validation is the de facto standard for model building and evaluation in machine learning, but care should be taken in adverse event prediction because this approach tends to be overoptimistic compared with the real-world situation. The time split, which uses the time axis, is considered suitable for real-world prediction. However, the differences in model performance obtained using the time and random splits are not fully understood. To understand the differences, we compared the model performance between the time and random splits using eight types of compound information as input, eight adverse events as targets, and six machine learning algorithms. The random split showed higher area under the curve values than did the time split for six of eight targets. The chemical spaces of the training and test datasets of the time split were similar, suggesting that the concept of applicability domain is insufficient to explain the differences derived from the splitting. The area under the curve differences were smaller for the protein interaction than for the other datasets. Subsequent detailed analyses suggested the danger of confounding in the use of knowledge-based information in the time split. These findings indicate the importance of understanding the differences between the time and random splits in adverse event prediction and suggest that appropriate use of the splitting strategies and interpretation of results are necessary for the real-world prediction of adverse events.