 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Attention Based Prioritization of Disease Responsible Genes from Multimodal Alzheimer's Network
Mar 01, 2026Prioritizing disease-associated genes is central to understanding the molecular mechanisms of complex disorders such as Alzheimer's disease (AD). Traditional network-based approaches rely on static centrality measures and often fail to capture cross-modal biological heterogeneity. We propose NETRA (Node Evaluation through Transformer-based Representation and Attention), a multimodal graph transformer framework that replaces heuristic centrality metrics with attention-driven relevance scoring. Using AD as a case study, gene regulatory networks are independently constructed from microarray, single-cell RNA-seq, and single-nucleus RNA-seq data. Random-walk sequences derived from these networks are used to train a BERT-based model for learning global gene embeddings, while modality-specific gene expression profiles are compressed using variational autoencoders. These representations are integrated with auxiliary biological networks, including protein-protein interactions, Gene Ontology semantic similarity, and diffusion-based gene similarity, into a unified multimodal graph. A graph transformer assigns NETRA scores that quantify gene relevance in a disease-specific and context-aware manner. Gene set enrichment analysis shows that NETRA achieves a normalized enrichment score of about 3.9 for the Alzheimer's disease pathway, substantially outperforming classical centrality measures and diffusion models. Top-ranked genes enrich multiple neurodegenerative pathways, recover a known late-onset AD susceptibility locus at chr12q13, and reveal conserved cross-disease gene modules. The framework preserves biologically realistic heavy-tailed network topology and is readily extensible to other complex disorders.
A Novel Graph Transformer Framework for Gene Regulatory Network Inference
Apr 23, 2025



The inference of gene regulatory networks (GRNs) is a foundational stride towards deciphering the fundamentals of complex biological systems. Inferring a possible regulatory link between two genes can be formulated as a link prediction problem. Inference of GRNs via gene coexpression profiling data may not always reflect true biological interactions, as its susceptibility to noise and misrepresenting true biological regulatory relationships. Most GRN inference methods face several challenges in the network reconstruction phase. Therefore, it is important to encode gene expression values, leverege the prior knowledge gained from the available inferred network structures and positional informations of the input network nodes towards inferring a better and more confident GRN network reconstruction. In this paper, we explore the integration of multiple inferred networks to enhance the inference of Gene Regulatory Networks (GRNs). Primarily, we employ autoencoder embeddings to capture gene expression patterns directly from raw data, preserving intricate biological signals. Then, we embed the prior knowledge from GRN structures transforming them into a text-like representation using random walks, which are then encoded with a masked language model, BERT, to generate global embeddings for each gene across all networks. Additionally, we embed the positional encodings of the input gene networks to better identify the position of each unique gene within the graph. These embeddings are integrated into graph transformer-based model, termed GT-GRN, for GRN inference. The GT-GRN model effectively utilizes the topological structure of the ground truth network while incorporating the enriched encoded information. Experimental results demonstrate that GT-GRN significantly outperforms existing GRN inference methods, achieving superior accuracy and highlighting the robustness of our approach.
Analyzing Host-Viral Interactome of SARS-CoV-2 for Identifying Vulnerable Host Proteins during COVID-19 Pathogenesis
Feb 05, 2021
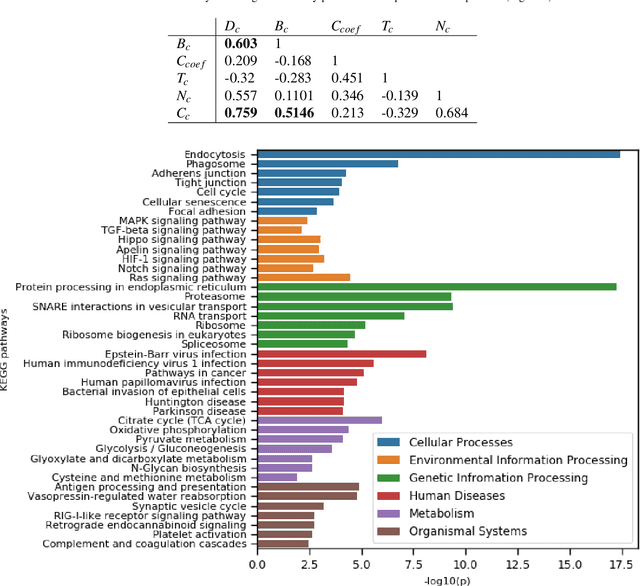
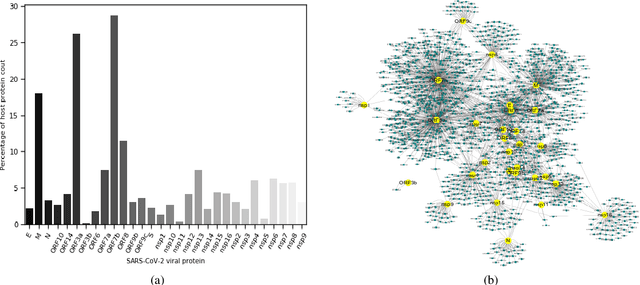

The development of therapeutic targets for COVID-19 treatment is based on the understanding of the molecular mechanism of pathogenesis. The identification of genes and proteins involved in the infection mechanism is the key to shed out light into the complex molecular mechanisms. The combined effort of many laboratories distributed throughout the world has produced the accumulation of both protein and genetic interactions. In this work we integrate these available results and we obtain an host protein-protein interaction network composed by 1432 human proteins. We calculate network centrality measures to identify key proteins. Then we perform functional enrichment of central proteins. We observed that the identified proteins are mostly associated with several crucial pathways, including cellular process, signalling transduction, neurodegenerative disease. Finally, we focused on proteins involved in causing disease in the human respiratory tract. We conclude that COVID19 is a complex disease, and we highlighted many potential therapeutic targets including RBX1, HSPA5, ITCH, RAB7A, RAB5A, RAB8A, PSMC5, CAPZB, CANX, IGF2R, HSPA1A, which are central and also associated with multiple diseases
Big Data Analytics in Bioinformatics: A Machine Learning Perspective
Jun 15, 2015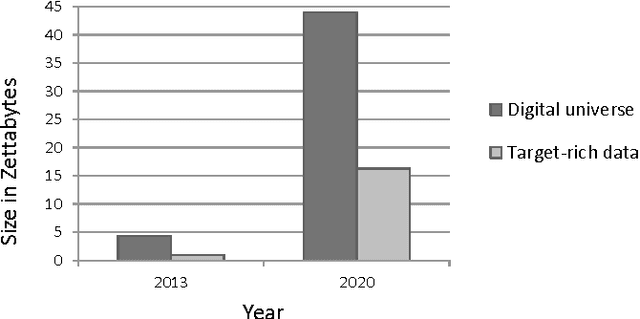

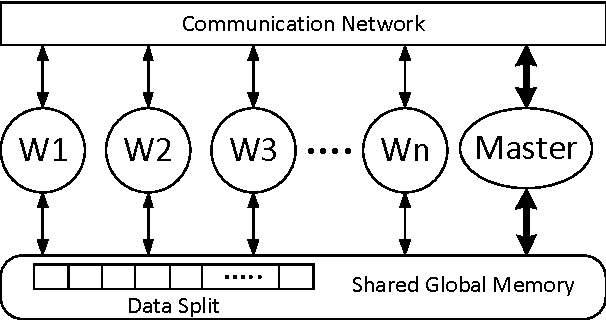

Bioinformatics research is characterized by voluminous and incremental datasets and complex data analytics methods. The machine learning methods used in bioinformatics are iterative and parallel. These methods can be scaled to handle big data using the distributed and parallel computing technologies. Usually big data tools perform computation in batch-mode and are not optimized for iterative processing and high data dependency among operations. In the recent years, parallel, incremental, and multi-view machine learning algorithms have been proposed. Similarly, graph-based architectures and in-memory big data tools have been developed to minimize I/O cost and optimize iterative processing. However, there lack standard big data architectures and tools for many important bioinformatics problems, such as fast construction of co-expression and regulatory networks and salient module identification, detection of complexes over growing protein-protein interaction data, fast analysis of massive DNA, RNA, and protein sequence data, and fast querying on incremental and heterogeneous disease networks. This paper addresses the issues and challenges posed by several big data problems in bioinformatics, and gives an overview of the state of the art and the future research opportunities.