 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLow-Dimensional High-Fidelity Kinetic Models for NOX Formation by a Compute Intensification Method
Feb 21, 2022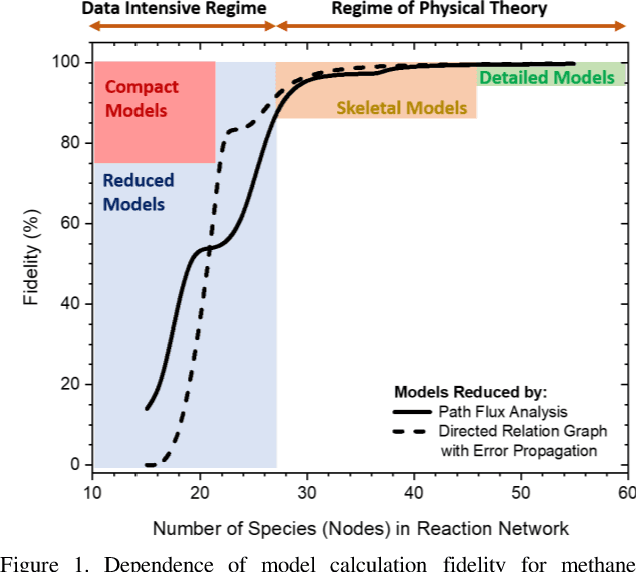
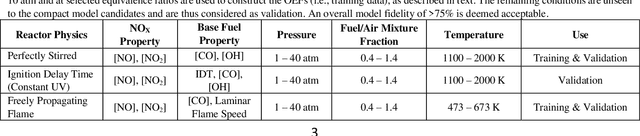
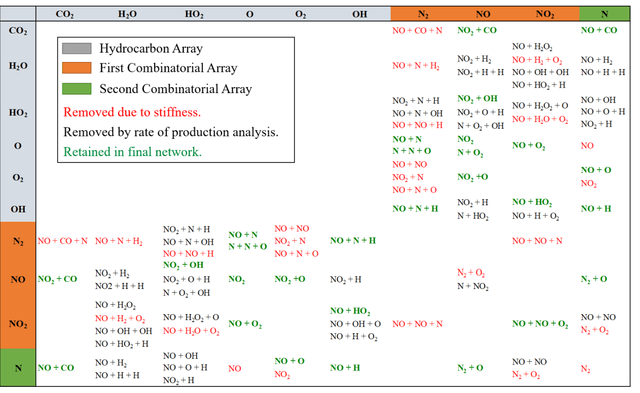
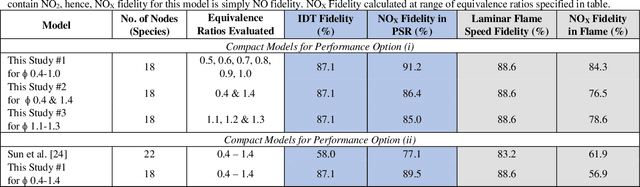
A novel compute intensification methodology to the construction of low-dimensional, high-fidelity "compact" kinetic models for NOX formation is designed and demonstrated. The method adapts the data intensive Machine Learned Optimization of Chemical Kinetics (MLOCK) algorithm for compact model generation by the use of a Latin Square method for virtual reaction network generation. A set of logical rules are defined which construct a minimally sized virtual reaction network comprising three additional nodes (N, NO, NO2). This NOX virtual reaction network is appended to a pre-existing compact model for methane combustion comprising fifteen nodes. The resulting eighteen node virtual reaction network is processed by the MLOCK coded algorithm to produce a plethora of compact model candidates for NOX formation during methane combustion. MLOCK automatically; populates the terms of the virtual reaction network with candidate inputs; measures the success of the resulting compact model candidates (in reproducing a broad set of gas turbine industry-defined performance targets); selects regions of input parameters space showing models of best performance; refines the input parameters to give better performance; and makes an ultimate selection of the best performing model or models. By this method, it is shown that a number of compact model candidates exist that show fidelities in excess of 75% in reproducing industry defined performance targets, with one model valid to >75% across fuel/air equivalence ratios of 0.5-1.0. However, to meet the full fuel/air equivalence ratio performance envelope defined by industry, we show that with this minimal virtual reaction network, two further compact models are required.
Toward Development of Machine Learned Techniques for Production of Compact Kinetic Models
Feb 16, 2022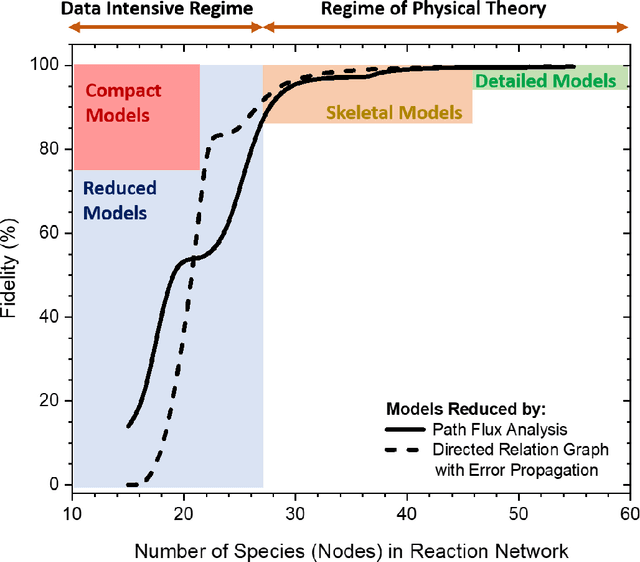
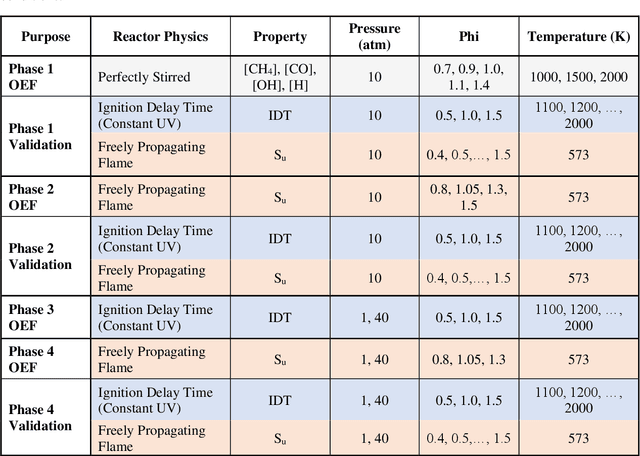
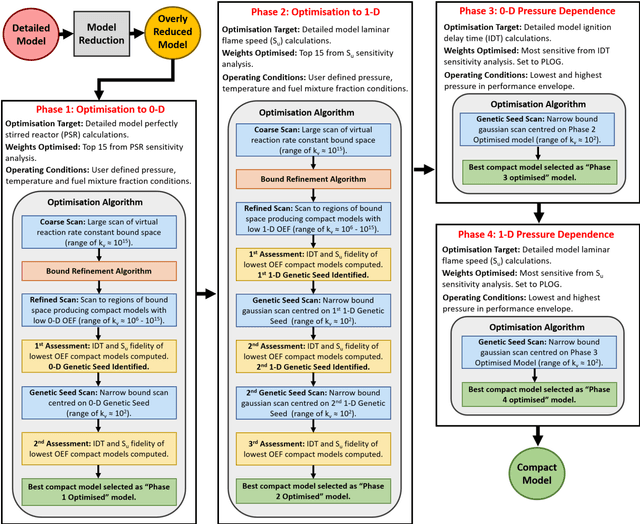
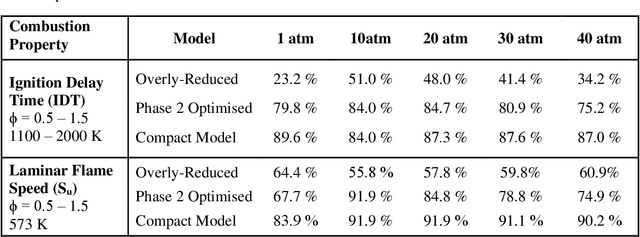
Chemical kinetic models are an essential component in the development and optimisation of combustion devices through their coupling to multi-dimensional simulations such as computational fluid dynamics (CFD). Low-dimensional kinetic models which retain good fidelity to the reality are needed, the production of which requires considerable human-time cost and expert knowledge. Here, we present a novel automated compute intensification methodology to produce overly-reduced and optimised (compact) chemical kinetic models. This algorithm, termed Machine Learned Optimisation of Chemical Kinetics (MLOCK), systematically perturbs each of the four sub-models of a chemical kinetic model to discover what combinations of terms results in a good model. A virtual reaction network comprised of n species is first obtained using conventional mechanism reduction. To counteract the imposed decrease in model performance, the weights (virtual reaction rate constants) of important connections (virtual reactions) between each node (species) of the virtual reaction network are numerically optimised to replicate selected calculations across four sequential phases. The first version of MLOCK, (MLOCK1.0) simultaneously perturbs all three virtual Arrhenius reaction rate constant parameters for important connections and assesses the suitability of the new parameters through objective error functions, which quantify the error in each compact model candidate's calculation of the optimisation targets, which may be comprised of detailed model calculations and/or experimental data. MLOCK1.0 is demonstrated by creating compact models for the archetypal case of methane air combustion. It is shown that the NUGMECH1.0 detailed model comprised of 2,789 species is reliably compacted to 15 species (nodes), whilst retaining an overall fidelity of ~87% to the detailed model calculations, outperforming the prior state-of-art.
Toward Machine Learned Highly Reduce Kinetic Models For Methane/Air Combustion
Mar 31, 2021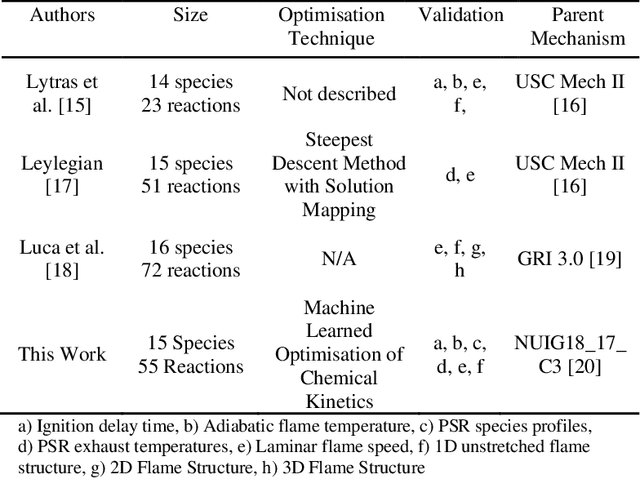
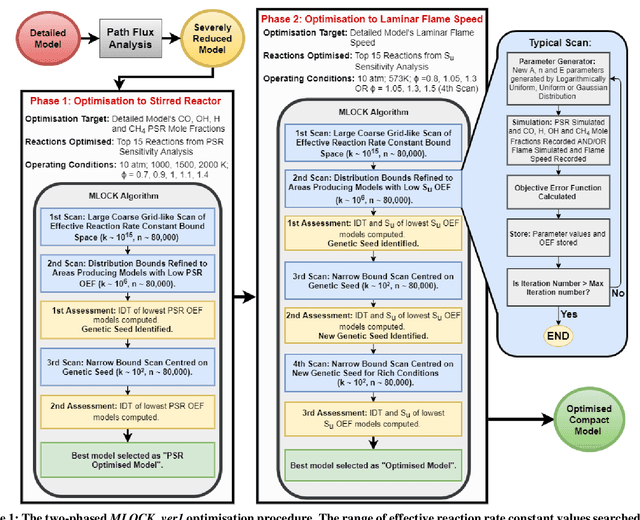
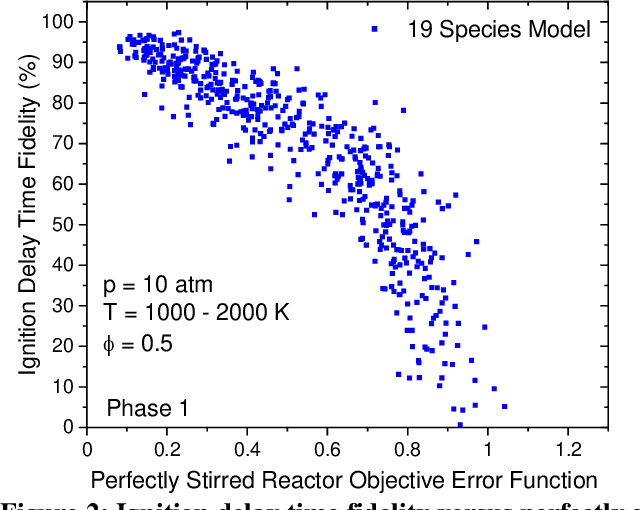
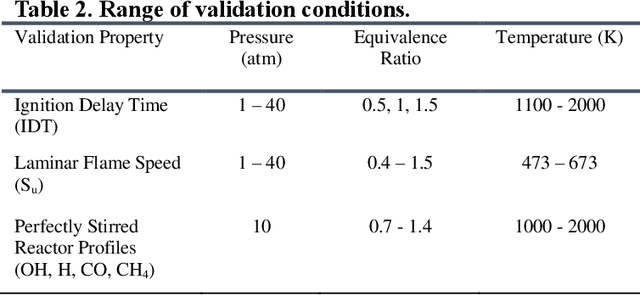
Accurate low dimension chemical kinetic models for methane are an essential component in the design of efficient gas turbine combustors. Kinetic models coupled to computational fluid dynamics (CFD) provide quick and efficient ways to test the effect of operating conditions, fuel composition and combustor design compared to physical experiments. However, detailed chemical kinetic models are too computationally expensive for use in CFD. We propose a novel data orientated three-step methodology to produce compact models that replicate a target set of detailed model properties to a high fidelity. In the first step, a reduced kinetic model is obtained by removing all non-essential species from the detailed model containing 118 species using path flux analysis (PFA). It is then numerically optimised to replicate the detailed model's prediction in two rounds; First, to selected species (OH,H,CO and CH4) profiles in perfectly stirred reactor (PSR) simulations and then re-optimised to the detailed model's prediction of the laminar flame speed. This is implemented by a purposely developed Machine Learned Optimisation of Chemical Kinetics (MLOCK) algorithm. The MLOCK algorithm systematically perturbs all three Arrhenius parameters for selected reactions and assesses the suitability of the new parameters through an objective error function which quantifies the error in the compact model's calculation of the optimisation target. This strategy is demonstrated through the production of a 19 species and a 15 species compact model for methane/air combustion. Both compact models are validated across a range of 0D and 1D calculations across both lean and rich conditions and shows good agreement to the parent detailed mechanism. The 15 species model is shown to outperform the current state-of-art models in both accuracy and range of conditions the model is valid over.