 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEffect of hyperparameters on variable selection in random forests
Sep 13, 2023Random forests (RFs) are well suited for prediction modeling and variable selection in high-dimensional omics studies. The effect of hyperparameters of the RF algorithm on prediction performance and variable importance estimation have previously been investigated. However, how hyperparameters impact RF-based variable selection remains unclear. We evaluate the effects on the Vita and the Boruta variable selection procedures based on two simulation studies utilizing theoretical distributions and empirical gene expression data. We assess the ability of the procedures to select important variables (sensitivity) while controlling the false discovery rate (FDR). Our results show that the proportion of splitting candidate variables (mtry.prop) and the sample fraction (sample.fraction) for the training dataset influence the selection procedures more than the drawing strategy of the training datasets and the minimal terminal node size. A suitable setting of the RF hyperparameters depends on the correlation structure in the data. For weakly correlated predictor variables, the default value of mtry is optimal, but smaller values of sample.fraction result in larger sensitivity. In contrast, the difference in sensitivity of the optimal compared to the default value of sample.fraction is negligible for strongly correlated predictor variables, whereas smaller values than the default are better in the other settings. In conclusion, the default values of the hyperparameters will not always be suitable for identifying important variables. Thus, adequate values differ depending on whether the aim of the study is optimizing prediction performance or variable selection.
Evaluation of network-guided random forest for disease gene discovery
Aug 02, 2023Gene network information is believed to be beneficial for disease module and pathway identification, but has not been explicitly utilized in the standard random forest (RF) algorithm for gene expression data analysis. We investigate the performance of a network-guided RF where the network information is summarized into a sampling probability of predictor variables which is further used in the construction of the RF. Our results suggest that network-guided RF does not provide better disease prediction than the standard RF. In terms of disease gene discovery, if disease genes form module(s), network-guided RF identifies them more accurately. In addition, when disease status is independent from genes in the given network, spurious gene selection results can occur when using network information, especially on hub genes. Our empirical analysis on two balanced microarray and RNA-Seq breast cancer datasets from The Cancer Genome Atlas (TCGA) for classification of progesterone receptor (PR) status also demonstrates that network-guided RF can identify genes from PGR-related pathways, which leads to a better connected module of identified genes.
A review on longitudinal data analysis with random forest in precision medicine
Aug 08, 2022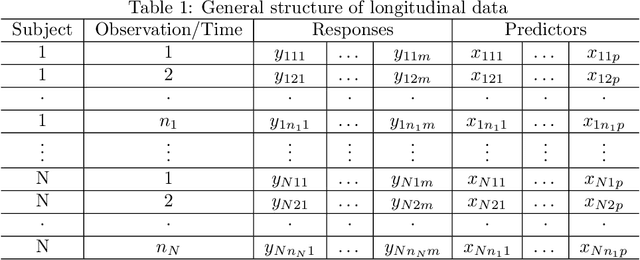
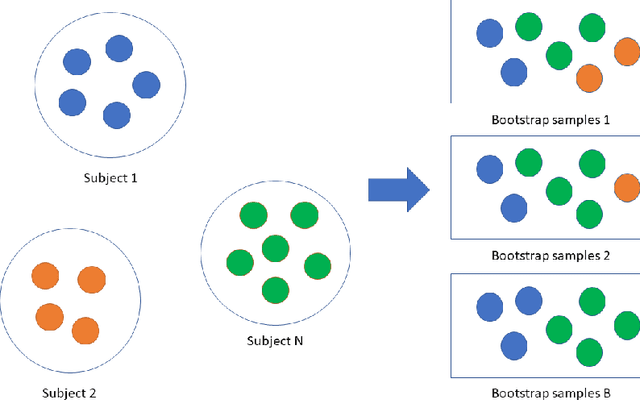
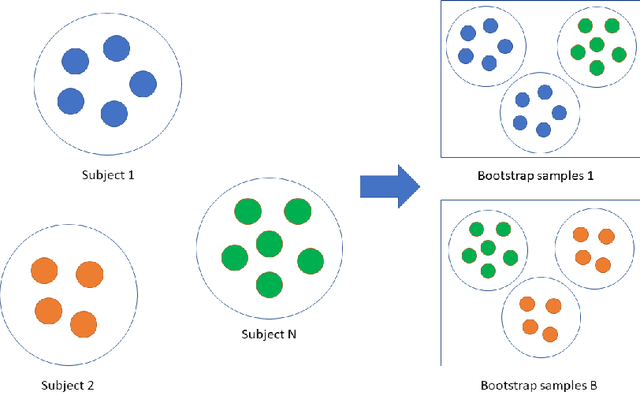

Precision medicine provides customized treatments to patients based on their characteristics and is a promising approach to improving treatment efficiency. Large scale omics data are useful for patient characterization, but often their measurements change over time, leading to longitudinal data. Random forest is one of the state-of-the-art machine learning methods for building prediction models, and can play a crucial role in precision medicine. In this paper, we review extensions of the standard random forest method for the purpose of longitudinal data analysis. Extension methods are categorized according to the data structures for which they are designed. We consider both univariate and multivariate responses and further categorize the repeated measurements according to whether the time effect is relevant. Information of available software implementations of the reviewed extensions is also given. We conclude with discussions on the limitations of our review and some future research directions.