 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Scalable Trie Building Algorithm for High-Throughput Phyloanalysis of Wafer-Scale Digital Evolution Experiments
Aug 20, 2025

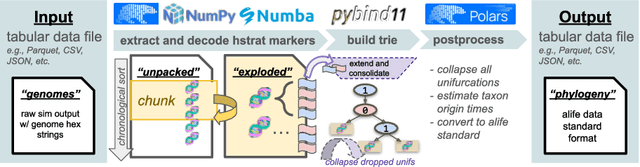
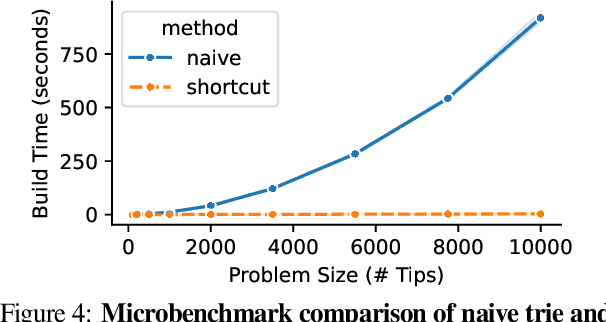
Agent-based simulation platforms play a key role in enabling fast-to-run evolution experiments that can be precisely controlled and observed in detail. Availability of high-resolution snapshots of lineage ancestries from digital experiments, in particular, is key to investigations of evolvability and open-ended evolution, as well as in providing a validation testbed for bioinformatics method development. Ongoing advances in AI/ML hardware accelerator devices, such as the 850,000-processor Cerebras Wafer-Scale Engine (WSE), are poised to broaden the scope of evolutionary questions that can be investigated in silico. However, constraints in memory capacity and locality characteristic of these systems introduce difficulties in exhaustively tracking phylogenies at runtime. To overcome these challenges, recent work on hereditary stratigraphy algorithms has developed space-efficient genetic markers to facilitate fully decentralized estimation of relatedness among digital organisms. However, in existing work, compute time to reconstruct phylogenies from these genetic markers has proven a limiting factor in achieving large-scale phyloanalyses. Here, we detail an improved trie-building algorithm designed to produce reconstructions equivalent to existing approaches. For modestly-sized 10,000-tip trees, the proposed approach achieves a 300-fold speedup versus existing state-of-the-art. Finally, using 1 billion genome datasets drawn from WSE simulations encompassing 954 trillion replication events, we report a pair of large-scale phylogeny reconstruction trials, achieving end-to-end reconstruction times of 2.6 and 2.9 hours. In substantially improving reconstruction scaling and throughput, presented work establishes a key foundation to enable powerful high-throughput phyloanalysis techniques in large-scale digital evolution experiments.
Extending a Phylogeny-based Method for Detecting Signatures of Multi-level Selection for Applications in Artificial Life
Aug 19, 2025Multilevel selection occurs when short-term individual-level reproductive interests conflict with longer-term group-level fitness effects. Detecting and quantifying this phenomenon is key to understanding evolution of traits ranging from multicellularity to pathogen virulence. Multilevel selection is particularly important in artificial life research due to its connection to major evolutionary transitions, a hallmark of open-ended evolution. Bonetti Franceschi & Volz (2024) proposed to detect multilevel selection dynamics by screening for mutations that appear more often in a population than expected by chance (due to individual-level fitness benefits) but are ultimately associated with negative longer-term fitness outcomes (i.e., smaller, shorter-lived descendant clades). Here, we use agent-based modeling with known ground truth to assess the efficacy of this approach. To test these methods under challenging conditions broadly comparable to the original dataset explored by Bonetti Franceschi & Volz (2024), we use an epidemiological framework to model multilevel selection in trade-offs between within-host growth rate and between-host transmissibility. To achieve success on our in silico data, we develop an alternate normalization procedure for identifying clade-level fitness effects. We find the method to be sensitive in detecting genome sites under multilevel selection with 30% effect sizes on fitness, but do not see sensitivity to smaller 10% mutation effect sizes. To test the robustness of this methodology, we conduct additional experiments incorporating extrinsic, time-varying environmental changes and adaptive turnover in population compositions, and find that screen performance remains generally consistent with baseline conditions. This work represents a promising step towards rigorous generalizable quantification of multilevel selection effects.
A Guide to Tracking Phylogenies in Parallel and Distributed Agent-based Evolution Models
May 16, 2024



Computer simulations are an important tool for studying the mechanics of biological evolution. In particular, in silico work with agent-based models provides an opportunity to collect high-quality records of ancestry relationships among simulated agents. Such phylogenies can provide insight into evolutionary dynamics within these simulations. Existing work generally tracks lineages directly, yielding an exact phylogenetic record of evolutionary history. However, direct tracking can be inefficient for large-scale, many-processor evolutionary simulations. An alternate approach to extracting phylogenetic information from simulation that scales more favorably is post hoc estimation, akin to how bioinformaticians build phylogenies by assessing genetic similarities between organisms. Recently introduced ``hereditary stratigraphy'' algorithms provide means for efficient inference of phylogenetic history from non-coding annotations on simulated organisms' genomes. A number of options exist in configuring hereditary stratigraphy methodology, but no work has yet tested how they impact reconstruction quality. To address this question, we surveyed reconstruction accuracy under alternate configurations across a matrix of evolutionary conditions varying in selection pressure, spatial structure, and ecological dynamics. We synthesize results from these experiments to suggest a prescriptive system of best practices for work with hereditary stratigraphy, ultimately guiding researchers in choosing appropriate instrumentation for large-scale simulation studies.
Phylotrack: C++ and Python libraries for in silico phylogenetic tracking
May 15, 2024

In silico evolution instantiates the processes of heredity, variation, and differential reproductive success (the three "ingredients" for evolution by natural selection) within digital populations of computational agents. Consequently, these populations undergo evolution, and can be used as virtual model systems for studying evolutionary dynamics. This experimental paradigm -- used across biological modeling, artificial life, and evolutionary computation -- complements research done using in vitro and in vivo systems by enabling experiments that would be impossible in the lab or field. One key benefit is complete, exact observability. For example, it is possible to perfectly record all parent-child relationships across simulation history, yielding complete phylogenies (ancestry trees). This information reveals when traits were gained or lost, and also facilitates inference of underlying evolutionary dynamics. The Phylotrack project provides libraries for tracking and analyzing phylogenies in in silico evolution. The project is composed of 1) Phylotracklib: a header-only C++ library, developed under the umbrella of the Empirical project, and 2) Phylotrackpy: a Python wrapper around Phylotracklib, created with Pybind11. Both components supply a public-facing API to attach phylogenetic tracking to digital evolution systems, as well as a stand-alone interface for measuring a variety of popular phylogenetic topology metrics. Underlying design and C++ implementation prioritizes efficiency, allowing for fast generational turnover for agent populations numbering in the tens of thousands. Several explicit features (e.g., phylogeny pruning and abstraction, etc.) are provided for reducing the memory footprint of phylogenetic information.
Case Study of Novelty, Complexity, and Adaptation in a Multicellular System
May 12, 2024



Continuing generation of novelty, complexity, and adaptation are well-established as core aspects of open-ended evolution. However, it has yet to be firmly established to what extent these phenomena are coupled and by what means they interact. In this work, we track the co-evolution of novelty, complexity, and adaptation in a case study from the DISHTINY simulation system, which is designed to study the evolution of digital multicellularity. In this case study, we describe ten qualitatively distinct multicellular morphologies, several of which exhibit asymmetrical growth and distinct life stages. We contextualize the evolutionary history of these morphologies with measurements of complexity and adaptation. Our case study suggests a loose -- sometimes divergent -- relationship can exist among novelty, complexity, and adaptation.
Ecology, Spatial Structure, and Selection Pressure Induce Strong Signatures in Phylogenetic Structure
May 12, 2024



Evolutionary dynamics are shaped by a variety of fundamental, generic drivers, including spatial structure, ecology, and selection pressure. These drivers impact the trajectory of evolution, and have been hypothesized to influence phylogenetic structure. Here, we set out to assess (1) if spatial structure, ecology, and selection pressure leave detectable signatures in phylogenetic structure, (2) the extent, in particular, to which ecology can be detected and discerned in the presence of spatial structure, and (3) the extent to which these phylogenetic signatures generalize across evolutionary systems. To this end, we analyze phylogenies generated by manipulating spatial structure, ecology, and selection pressure within three computational models of varied scope and sophistication. We find that selection pressure, spatial structure, and ecology have characteristic effects on phylogenetic metrics, although these effects are complex and not always intuitive. Signatures have some consistency across systems when using equivalent taxonomic unit definitions (e.g., individual, genotype, species). Further, we find that sufficiently strong ecology can be detected in the presence of spatial structure. We also find that, while low-resolution phylogenetic reconstructions can bias some phylogenetic metrics, high-resolution reconstructions recapitulate them faithfully. Although our results suggest potential for evolutionary inference of spatial structure, ecology, and selection pressure through phylogenetic analysis, further methods development is needed to distinguish these drivers' phylometric signatures from each other and to appropriately normalize phylogenetic metrics. With such work, phylogenetic analysis could provide a versatile toolkit to study large-scale evolving populations.
Trackable Island-model Genetic Algorithms at Wafer Scale
May 06, 2024

Emerging ML/AI hardware accelerators, like the 850,000 processor Cerebras Wafer-Scale Engine (WSE), hold great promise to scale up the capabilities of evolutionary computation. However, challenges remain in maintaining visibility into underlying evolutionary processes while efficiently utilizing these platforms' large processor counts. Here, we focus on the problem of extracting phylogenetic information from digital evolution on the WSE platform. We present a tracking-enabled asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations a minute for population sizes reaching 16 million. This pace enables quadrillions of evaluations a day. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction of clear phylometric signals that differentiate wafer-scale runs with adaptive dynamics enabled versus disabled. Together, these benchmark and validation trials reflect strong potential for highly scalable evolutionary computation that is both efficient and observable. Kernel code implementing the island-model GA supports drop-in customization to support any fixed-length genome content and fitness criteria, allowing it to be leveraged to advance research interests across the community.
Methods to Estimate Cryptic Sequence Complexity
Apr 16, 2024
Complexity is a signature quality of interest in artificial life systems. Alongside other dimensions of assessment, it is common to quantify genome sites that contribute to fitness as a complexity measure. However, limitations to the sensitivity of fitness assays in models with implicit replication criteria involving rich biotic interactions introduce the possibility of difficult-to-detect ``cryptic'' adaptive sites, which contribute small fitness effects below the threshold of individual detectability or involve epistatic redundancies. Here, we propose three knockout-based assay procedures designed to quantify cryptic adaptive sites within digital genomes. We report initial tests of these methods on a simple genome model with explicitly configured site fitness effects. In these limited tests, estimation results reflect ground truth cryptic sequence complexities well. Presented work provides initial steps toward development of new methods and software tools that improve the resolution, rigor, and tractability of complexity analyses across alife systems, particularly those requiring expensive in situ assessments of organism fitness.
Trackable Agent-based Evolution Models at Wafer Scale
Apr 16, 2024



Continuing improvements in computing hardware are poised to transform capabilities for in silico modeling of cross-scale phenomena underlying major open questions in evolutionary biology and artificial life, such as transitions in individuality, eco-evolutionary dynamics, and rare evolutionary events. Emerging ML/AI-oriented hardware accelerators, like the 850,000 processor Cerebras Wafer Scale Engine (WSE), hold particular promise. However, practical challenges remain in conducting informative evolution experiments that efficiently utilize these platforms' large processor counts. Here, we focus on the problem of extracting phylogenetic information from agent-based evolution on the WSE platform. This goal drove significant refinements to decentralized in silico phylogenetic tracking, reported here. These improvements yield order-of-magnitude performance improvements. We also present an asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations a minute for population sizes reaching 16 million agents. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction, from wafer-scale simulation, of clear phylometric signals that differentiate runs with adaptive dynamics enabled versus disabled. Together, these benchmark and validation trials reflect strong potential for highly scalable agent-based evolution simulation that is both efficient and observable. Developed capabilities will bring entirely new classes of previously intractable research questions within reach, benefiting further explorations within the evolutionary biology and artificial life communities across a variety of emerging high-performance computing platforms.
Runtime phylogenetic analysis enables extreme subsampling for test-based problems
Feb 02, 2024



A phylogeny describes the evolutionary history of an evolving population. Evolutionary search algorithms can perfectly track the ancestry of candidate solutions, illuminating a population's trajectory through the search space. However, phylogenetic analyses are typically limited to post-hoc studies of search performance. We introduce phylogeny-informed subsampling, a new class of subsampling methods that exploit runtime phylogenetic analyses for solving test-based problems. Specifically, we assess two phylogeny-informed subsampling methods -- individualized random subsampling and ancestor-based subsampling -- on three diagnostic problems and ten genetic programming (GP) problems from program synthesis benchmark suites. Overall, we found that phylogeny-informed subsampling methods enable problem-solving success at extreme subsampling levels where other subsampling methods fail. For example, phylogeny-informed subsampling methods more reliably solved program synthesis problems when evaluating just one training case per-individual, per-generation. However, at moderate subsampling levels, phylogeny-informed subsampling generally performed no better than random subsampling on GP problems. Our diagnostic experiments show that phylogeny-informed subsampling improves diversity maintenance relative to random subsampling, but its effects on a selection scheme's capacity to rapidly exploit fitness gradients varied by selection scheme. Continued refinements of phylogeny-informed subsampling techniques offer a promising new direction for scaling up evolutionary systems to handle problems with many expensive-to-evaluate fitness criteria.