 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFeature Importance Explanations for Temporal Black-Box Models
Feb 23, 2021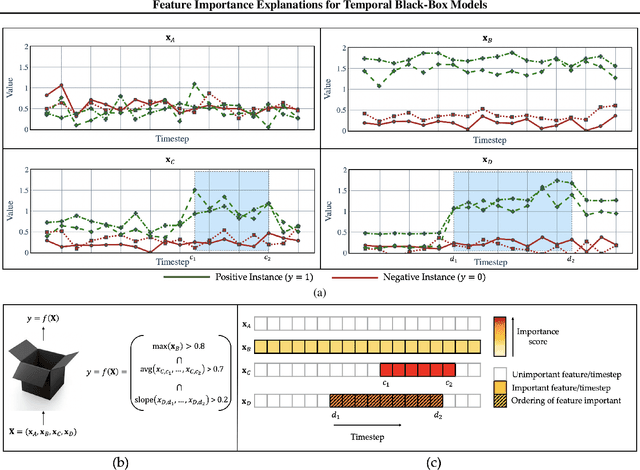
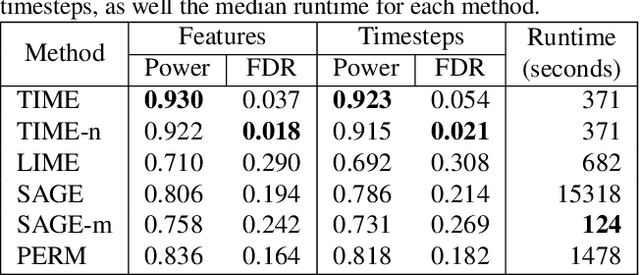
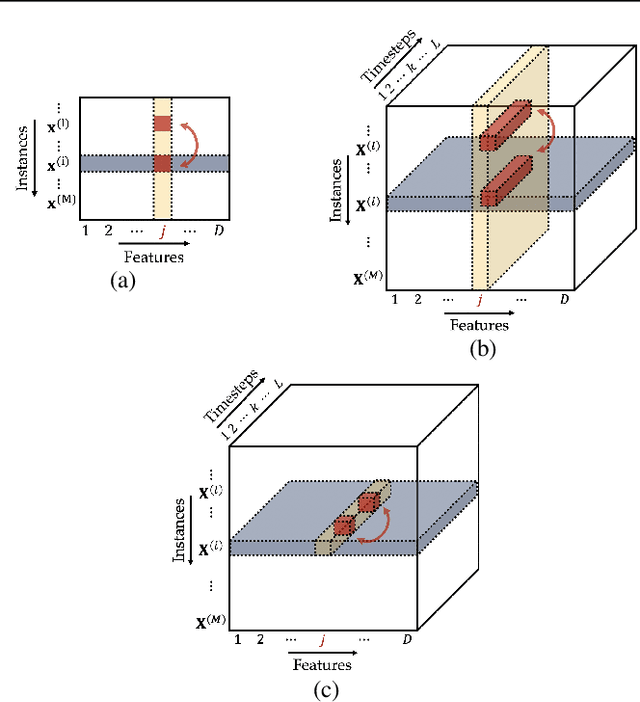
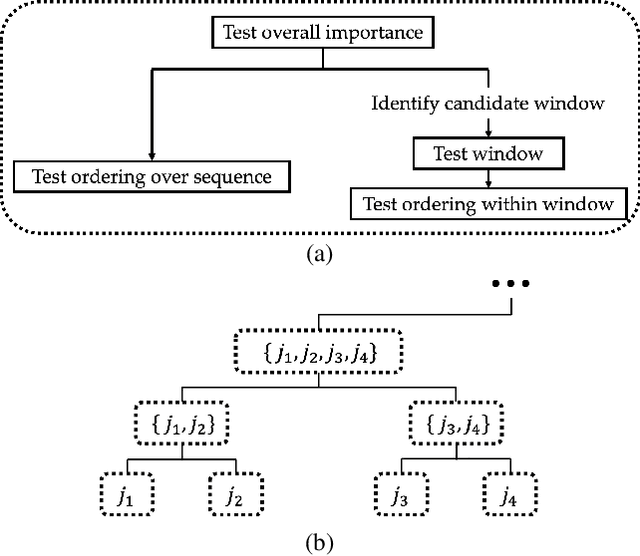
Models in the supervised learning framework may capture rich and complex representations over the features that are hard for humans to interpret. Existing methods to explain such models are often specific to architectures and data where the features do not have a time-varying component. In this work, we propose TIME, a method to explain models that are inherently temporal in nature. Our approach (i) uses a model-agnostic permutation-based approach to analyze global feature importance, (ii) identifies the importance of salient features with respect to their temporal ordering as well as localized windows of influence, and (iii) uses hypothesis testing to provide statistical rigor.
Understanding Learned Models by Identifying Important Features at the Right Resolution
Nov 20, 2018

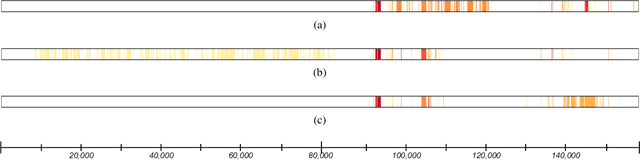
In many application domains, it is important to characterize how complex learned models make their decisions across the distribution of instances. One way to do this is to identify the features and interactions among them that contribute to a model's predictive accuracy. We present a model-agnostic approach to this task that makes the following specific contributions. Our approach (i) tests feature groups, in addition to base features, and tries to determine the level of resolution at which important features can be determined, (ii) uses hypothesis testing to rigorously assess the effect of each feature on the model's loss, (iii) employs a hierarchical approach to control the false discovery rate when testing feature groups and individual base features for importance, and (iv) uses hypothesis testing to identify important interactions among features and feature groups. We evaluate our approach by analyzing random forest and LSTM neural network models learned in two challenging biomedical applications.
Learning Hidden Markov Models for Regression using Path Aggregation
Jun 13, 2012
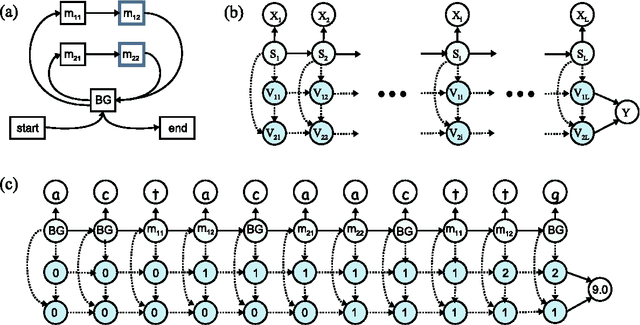
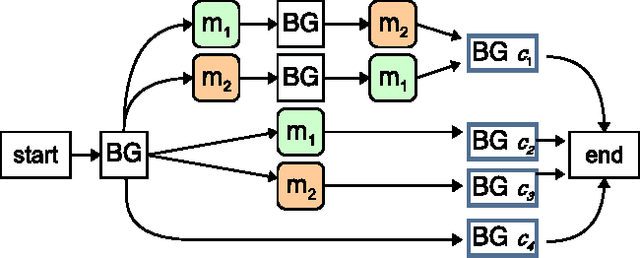
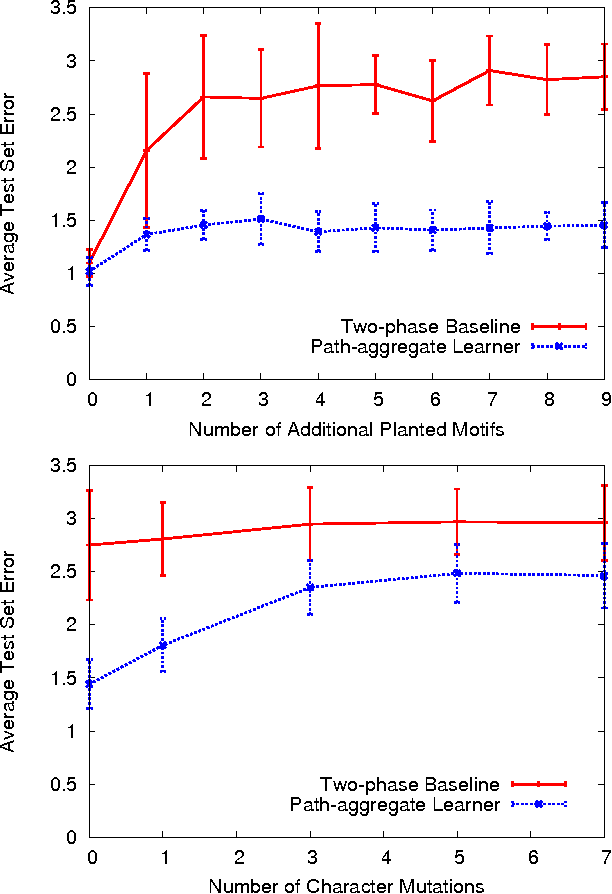
We consider the task of learning mappings from sequential data to real-valued responses. We present and evaluate an approach to learning a type of hidden Markov model (HMM) for regression. The learning process involves inferring the structure and parameters of a conventional HMM, while simultaneously learning a regression model that maps features that characterize paths through the model to continuous responses. Our results, in both synthetic and biological domains, demonstrate the value of jointly learning the two components of our approach.