 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApplication of generative autoencoder in de novo molecular design
Nov 21, 2017
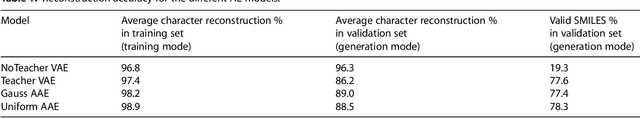
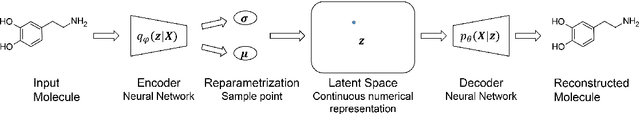

A major challenge in computational chemistry is the generation of novel molecular structures with desirable pharmacological and physiochemical properties. In this work, we investigate the potential use of autoencoder, a deep learning methodology, for de novo molecular design. Various generative autoencoders were used to map molecule structures into a continuous latent space and vice versa and their performance as structure generator was assessed. Our results show that the latent space preserves chemical similarity principle and thus can be used for the generation of analogue structures. Furthermore, the latent space created by autoencoders were searched systematically to generate novel compounds with predicted activity against dopamine receptor type 2 and compounds similar to known active compounds not included in the training set were identified.
Molecular De Novo Design through Deep Reinforcement Learning
Aug 29, 2017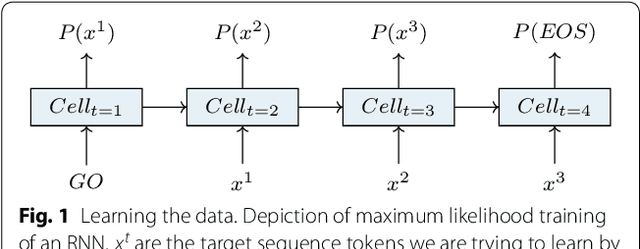
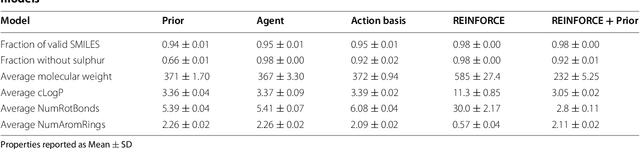
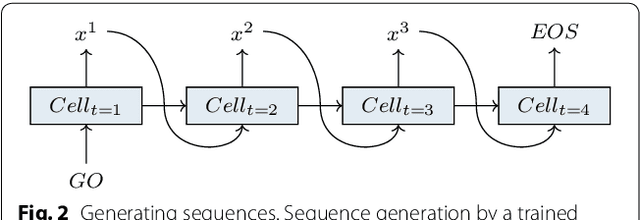

This work introduces a method to tune a sequence-based generative model for molecular de novo design that through augmented episodic likelihood can learn to generate structures with certain specified desirable properties. We demonstrate how this model can execute a range of tasks such as generating analogues to a query structure and generating compounds predicted to be active against a biological target. As a proof of principle, the model is first trained to generate molecules that do not contain sulphur. As a second example, the model is trained to generate analogues to the drug Celecoxib, a technique that could be used for scaffold hopping or library expansion starting from a single molecule. Finally, when tuning the model towards generating compounds predicted to be active against the dopamine receptor type 2, the model generates structures of which more than 95% are predicted to be active, including experimentally confirmed actives that have not been included in either the generative model nor the activity prediction model.