 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFree-rider Attacks on Model Aggregation in Federated Learning
Jun 21, 2020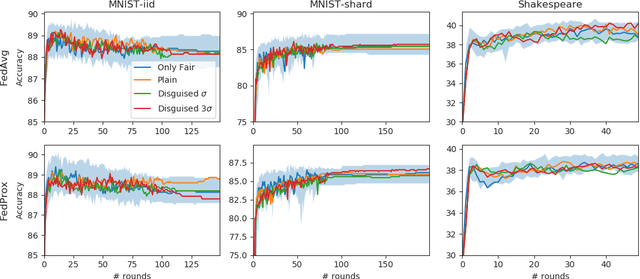
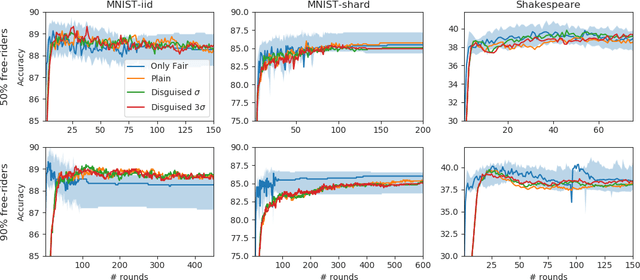
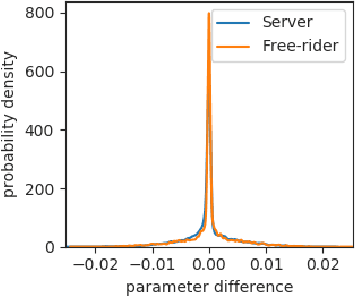
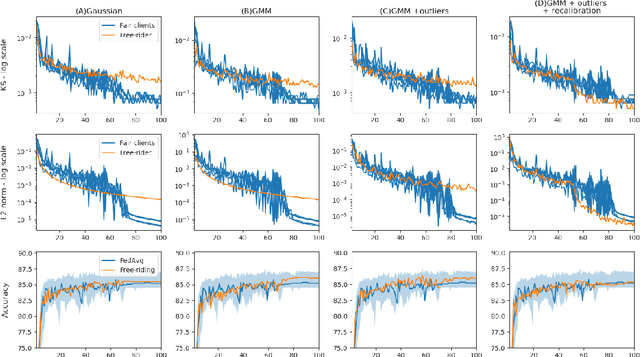
Free-rider attacks on federated learning consist in dissimulating participation to the federated learning process with the goal of obtaining the final aggregated model without actually contributing with any data. We introduce here the first theoretical and experimental analysis of free-rider attacks on federated learning schemes based on iterative parameters aggregation, such as FedAvg or FedProx, and provide formal guarantees for these attacks to converge to the aggregated models of the fair participants. We first show that a straightforward implementation of this attack can be simply achieved by not updating the local parameters during the iterative federated optimization. As this attack can be detected by adopting simple countermeasures at the server level, we subsequently study more complex disguising schemes based on stochastic updates of the free-rider parameters. We demonstrate the proposed strategies on a number of experimental scenarios, in both iid and non-iid settings. We conclude by providing recommendations to avoid free-rider attacks in real world applications of federated learning, especially in sensitive domains where security of data and models is critical.
A model of brain morphological changes related to aging and Alzheimer's disease from cross-sectional assessments
May 23, 2019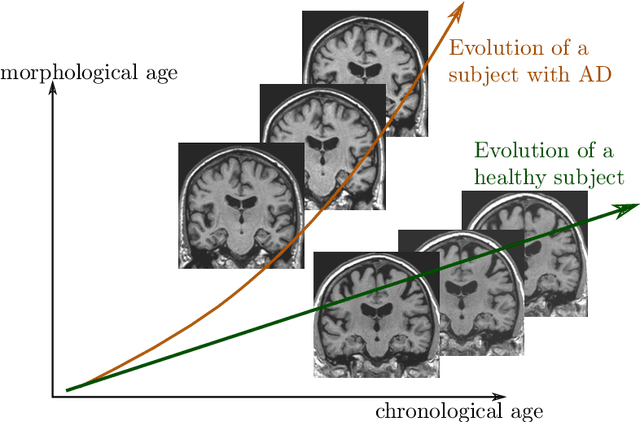
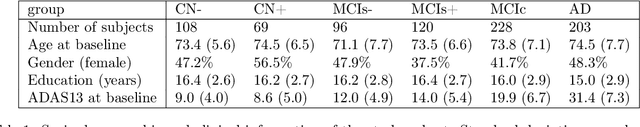
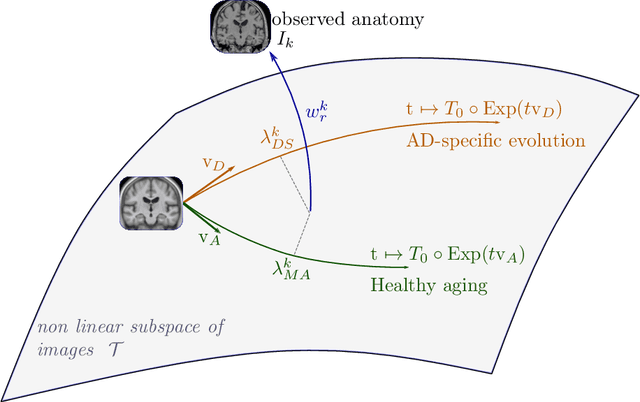

In this study we propose a deformation-based framework to jointly model the influence of aging and Alzheimer's disease (AD) on the brain morphological evolution. Our approach combines a spatio-temporal description of both processes into a generative model. A reference morphology is deformed along specific trajectories to match subject specific morphologies. It is used to define two imaging progression markers: 1) a morphological age and 2) a disease score. These markers can be computed locally in any brain region. The approach is evaluated on brain structural magnetic resonance images (MRI) from the ADNI database. The generative model is first estimated on a control population, then, for each subject, the markers are computed for each acquisition. The longitudinal evolution of these markers is then studied in relation with the clinical diagnosis of the subjects and used to generate possible morphological evolution. In the model, the morphological changes associated with normal aging are mainly found around the ventricles, while the Alzheimer's disease specific changes are more located in the temporal lobe and the hippocampal area. The statistical analysis of these markers highlights differences between clinical conditions even though the inter-subject variability is quiet high. In this context, the model can be used to generate plausible morphological trajectories associated with the disease. Our method gives two interpretable scalar imaging biomarkers assessing the effects of aging and disease on brain morphology at the individual and population level. These markers confirm an acceleration of apparent aging for Alzheimer's subjects and can help discriminate clinical conditions even in prodromal stages. More generally, the joint modeling of normal and pathological evolutions shows promising results to describe age-related brain diseases over long time scales.
Monotonic Gaussian Process for Spatio-Temporal Trajectory Separation in Brain Imaging Data
Feb 28, 2019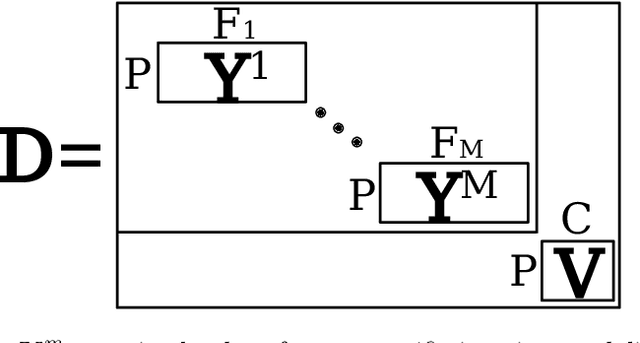

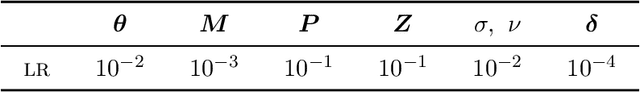
We introduce a probabilistic generative model for disentangling spatio-temporal disease trajectories from series of high-dimensional brain images. The model is based on spatio-temporal matrix factorization, where inference on the sources is constrained by anatomically plausible statistical priors. To model realistic trajectories, the temporal sources are defined as monotonic and time-reparametrized Gaussian Processes. To account for the non-stationarity of brain images, we model the spatial sources as sparse codes convolved at multiple scales. The method was tested on synthetic data favourably comparing with standard blind source separation approaches. The application on large-scale imaging data from a clinical study allows to disentangle differential temporal progression patterns mapping brain regions key to neurodegeneration, while revealing a disease-specific time scale associated to the clinical diagnosis.
DIVE: A spatiotemporal progression model of brain pathology in neurodegenerative disorders
Jan 11, 2019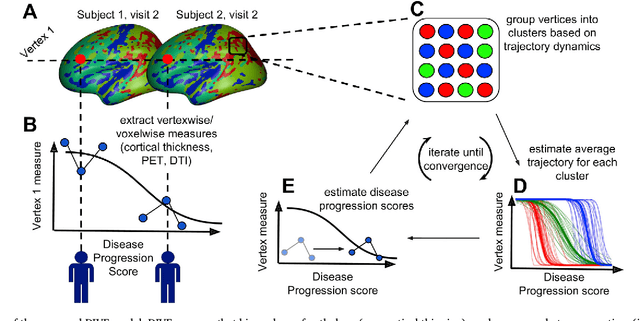
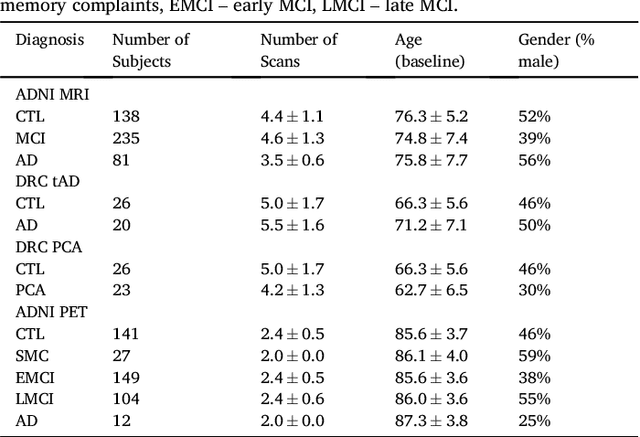
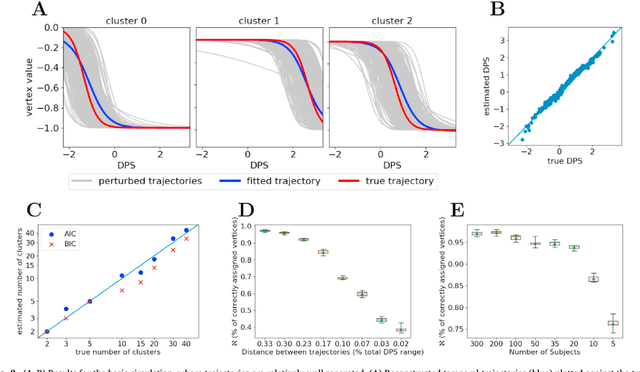
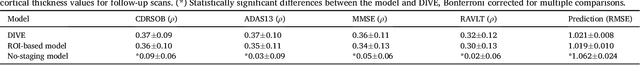
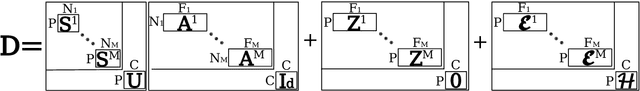
Here we present DIVE: Data-driven Inference of Vertexwise Evolution. DIVE is an image-based disease progression model with single-vertex resolution, designed to reconstruct long-term patterns of brain pathology from short-term longitudinal data sets. DIVE clusters vertex-wise biomarker measurements on the cortical surface that have similar temporal dynamics across a patient population, and concurrently estimates an average trajectory of vertex measurements in each cluster. DIVE uniquely outputs a parcellation of the cortex into areas with common progression patterns, leading to a new signature for individual diseases. DIVE further estimates the disease stage and progression speed for every visit of every subject, potentially enhancing stratification for clinical trials or management. On simulated data, DIVE can recover ground truth clusters and their underlying trajectory, provided the average trajectories are sufficiently different between clusters. We demonstrate DIVE on data from two cohorts: the Alzheimer's Disease Neuroimaging Initiative (ADNI) and the Dementia Research Centre (DRC), UK, containing patients with Posterior Cortical Atrophy (PCA) as well as typical Alzheimer's disease (tAD). DIVE finds similar spatial patterns of atrophy for tAD subjects in the two independent datasets (ADNI and DRC), and further reveals distinct patterns of pathology in different diseases (tAD vs PCA) and for distinct types of biomarker data: cortical thickness from Magnetic Resonance Imaging (MRI) vs amyloid load from Positron Emission Tomography (PET). Finally, DIVE can be used to estimate a fine-grained spatial distribution of pathology in the brain using any kind of voxelwise or vertexwise measures including Jacobian compression maps, fractional anisotropy (FA) maps from diffusion imaging or other PET measures. DIVE source code is available online: https://github.com/mrazvan22/dive
Disease Knowledge Transfer across Neurodegenerative Diseases
Jan 11, 2019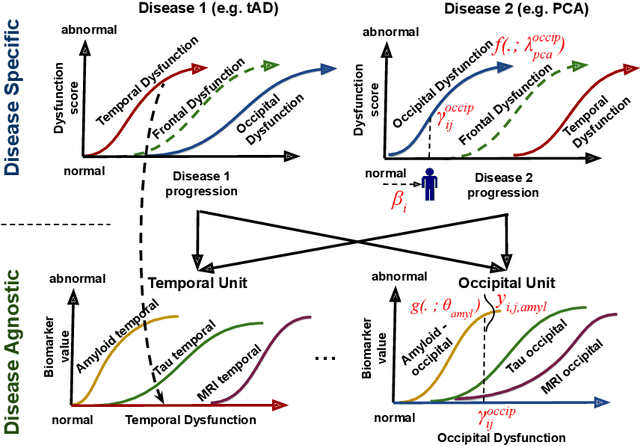
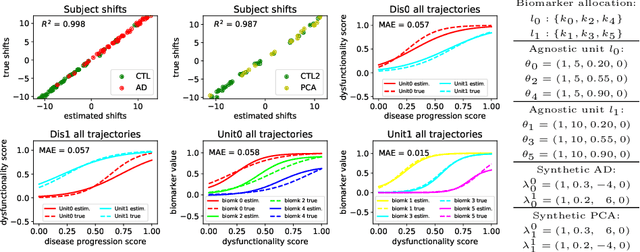
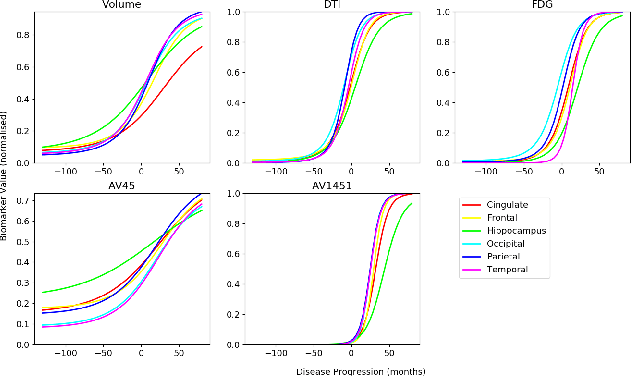

We introduce Disease Knowledge Transfer (DKT), a novel technique for transferring biomarker information between related neurodegenerative diseases. DKT infers robust multimodal biomarker trajectories in rare neurodegenerative diseases even when only limited, unimodal data is available, by transferring information from larger multimodal datasets from common neurodegenerative diseases. DKT is a joint-disease generative model of biomarker progressions, which exploits biomarker relationships that are shared across diseases. As opposed to current deep learning approaches, DKT is interpretable, which allows us to understand underlying disease mechanisms. Here we demonstrate DKT on Alzheimer's disease (AD) variants and its ability to predict trajectories for multimodal biomarkers in Posterior Cortical Atrophy (PCA), in lack of such data from PCA subjects. For this we train DKT on a combined dataset containing subjects with two distinct diseases and sizes of data available: 1) a larger, multimodal typical AD (tAD) dataset from the TADPOLE Challenge, and 2) a smaller unimodal Posterior Cortical Atrophy (PCA) dataset from the Dementia Research Centre (DRC) UK, for which only a limited number of Magnetic Resonance Imaging (MRI) scans are available. We first show that DKT estimates plausible multimodal trajectories in PCA that agree with previous literature. We further validate DKT in two situations: (1) on synthetic data, showing that it can accurately estimate the ground truth parameters and (2) on 20 DTI scans from controls and PCA patients, showing that it has favourable predictive performance compared to standard approaches. While we demonstrated DKT on Alzheimer's variants, we note DKT is generalisable to other forms of related neurodegenerative diseases. Source code for DKT is available online: https://github.com/mrazvan22/dkt.
Federated Learning in Distributed Medical Databases: Meta-Analysis of Large-Scale Subcortical Brain Data
Oct 22, 2018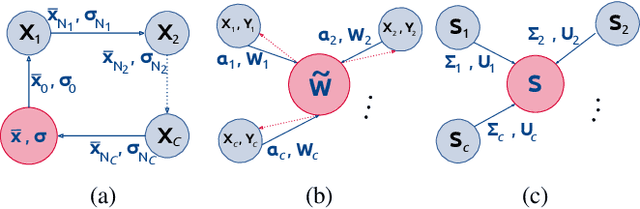

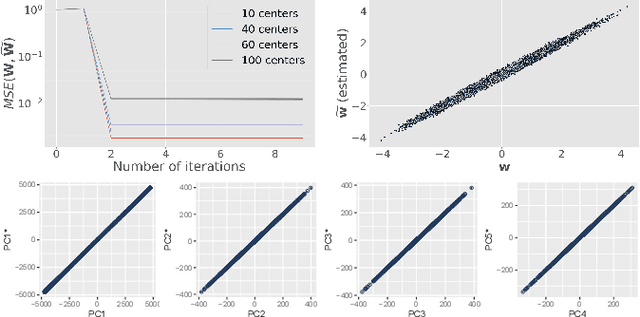
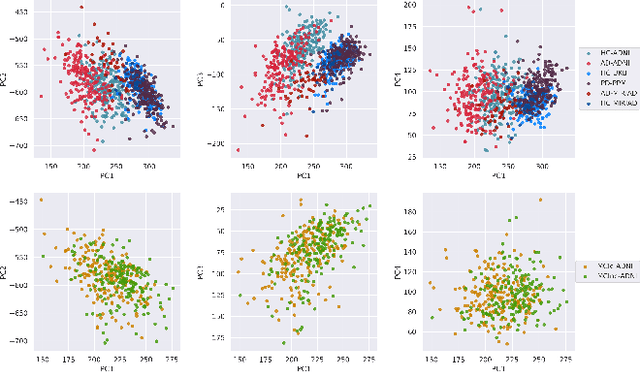
At this moment, databanks worldwide contain brain images of previously unimaginable numbers. Combined with developments in data science, these massive data provide the potential to better understand the genetic underpinnings of brain diseases. However, different datasets, which are stored at different institutions, cannot always be shared directly due to privacy and legal concerns, thus limiting the full exploitation of big data in the study of brain disorders. Here we propose a federated learning framework for securely accessing and meta-analyzing any biomedical data without sharing individual information. We illustrate our framework by investigating brain structural relationships across diseases and clinical cohorts. The framework is first tested on synthetic data and then applied to multi-centric, multi-database studies including ADNI, PPMI, MIRIAD and UK Biobank, showing the potential of the approach for further applications in distributed analysis of multi-centric cohorts
Constraining the Dynamics of Deep Probabilistic Models
Jun 18, 2018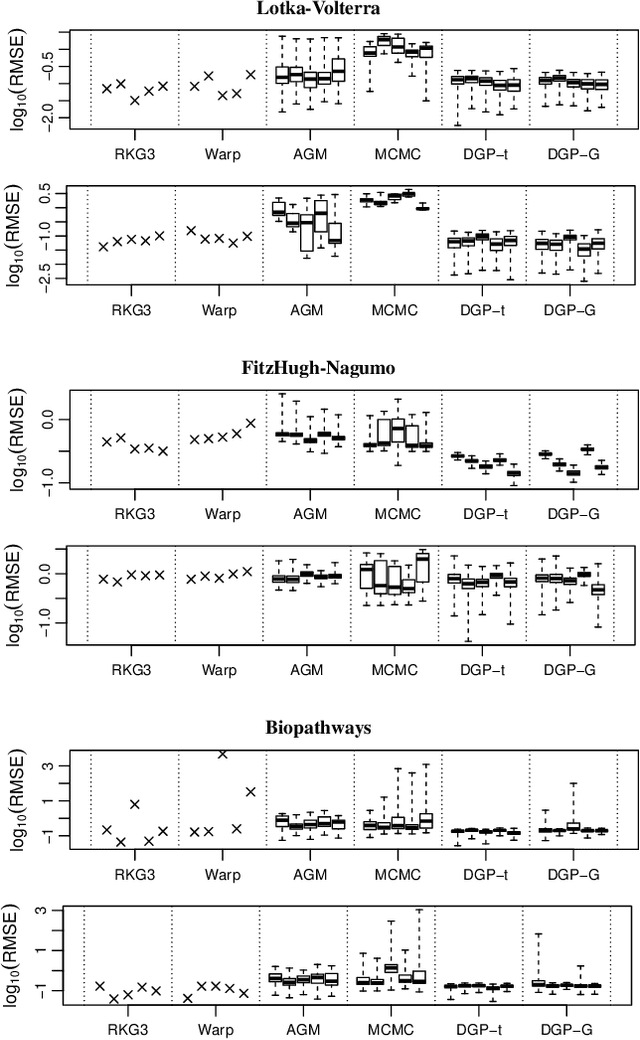
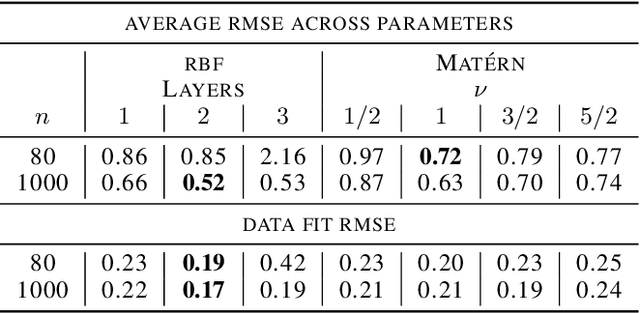
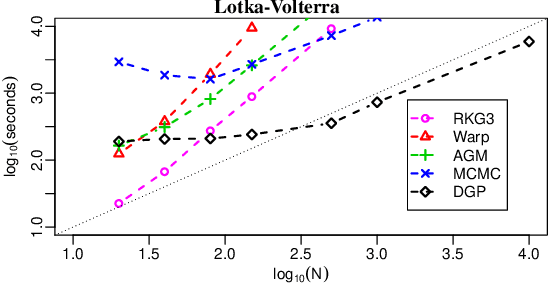
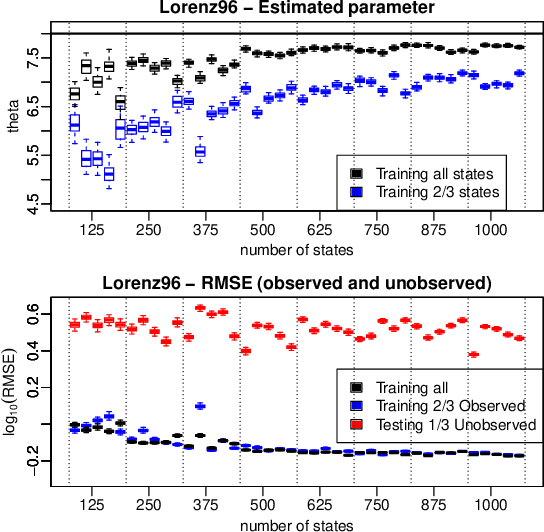
We introduce a novel generative formulation of deep probabilistic models implementing "soft" constraints on their function dynamics. In particular, we develop a flexible methodological framework where the modeled functions and derivatives of a given order are subject to inequality or equality constraints. We then characterize the posterior distribution over model and constraint parameters through stochastic variational inference. As a result, the proposed approach allows for accurate and scalable uncertainty quantification on the predictions and on all parameters. We demonstrate the application of equality constraints in the challenging problem of parameter inference in ordinary differential equation models, while we showcase the application of inequality constraints on the problem of monotonic regression of count data. The proposed approach is extensively tested in several experimental settings, leading to highly competitive results in challenging modeling applications, while offering high expressiveness, flexibility and scalability.