 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA novel and effective scoring scheme for structure classification and pairwise similarity measurement
Oct 04, 2016
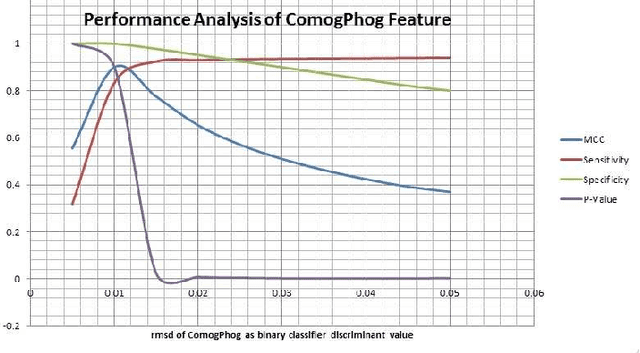
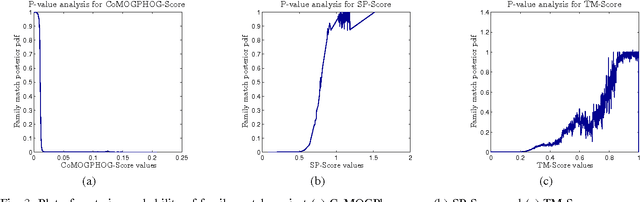
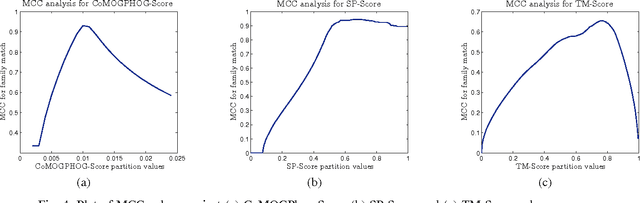
Protein tertiary structure defines its functions, classification and binding sites. Similar structural characteristics between two proteins often lead to the similar characteristics thereof. Determining structural similarity accurately in real time is a crucial research issue. In this paper, we present a novel and effective scoring scheme that is dependent on novel features extracted from protein alpha carbon distance matrices. Our scoring scheme is inspired from pattern recognition and computer vision. Our method is significantly better than the current state of the art methods in terms of family match of pairs of protein structures and other statistical measurements. The effectiveness of our method is tested on standard benchmark structures. A web service is available at http://research.buet.ac.bd:8080/Comograd/score.html where you can get the similarity measurement score between two protein structures based on our method.
GreMuTRRR: A Novel Genetic Algorithm to Solve Distance Geometry Problem for Protein Structures
Nov 16, 2014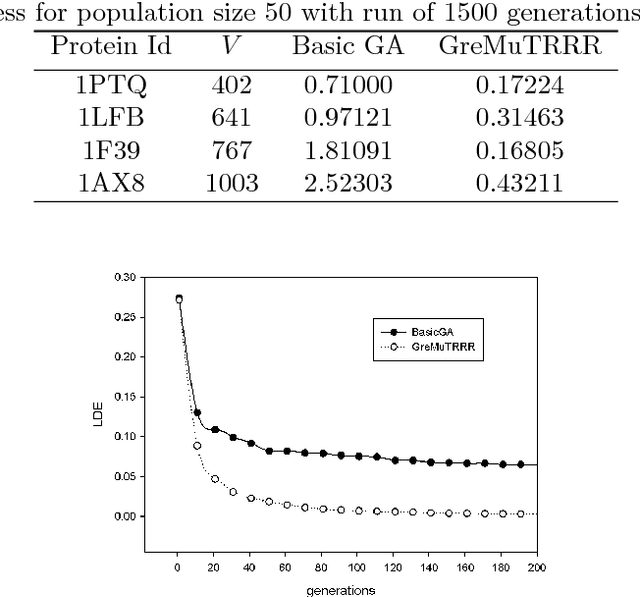
Nuclear Magnetic Resonance (NMR) Spectroscopy is a widely used technique to predict the native structure of proteins. However, NMR machines are only able to report approximate and partial distances between pair of atoms. To build the protein structure one has to solve the Euclidean distance geometry problem given the incomplete interval distance data produced by NMR machines. In this paper, we propose a new genetic algorithm for solving the Euclidean distance geometry problem for protein structure prediction given sparse NMR data. Our genetic algorithm uses a greedy mutation operator to intensify the search, a twin removal technique for diversification in the population and a random restart method to recover stagnation. On a standard set of benchmark dataset, our algorithm significantly outperforms standard genetic algorithms.
CoMOGrad and PHOG: From Computer Vision to Fast and Accurate Protein Tertiary Structure Retrieval
Sep 02, 2014


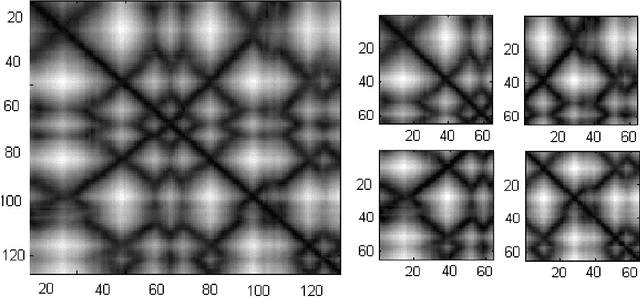
Due to the advancements in technology number of entries in the structural database of proteins are increasing day by day. Methods for retrieving protein tertiary structures from this large database is the key to comparative analysis of structures which plays an important role to understand proteins and their function. In this paper, we present fast and accurate methods for the retrieval of proteins from a large database with tertiary structures similar to a query protein. Our proposed methods borrow ideas from the field of computer vision. The speed and accuracy of our methods comes from the two newly introduced features, the co-occurrence matrix of the oriented gradient and pyramid histogram of oriented gradient and from the use of Euclidean distance as the distance measure. Experimental results clearly indicate the superiority of our approach in both running time and accuracy. Our method is readily available for use from this website: http://research.buet.ac.bd:8080/Comograd/.
Solving the Minimum Common String Partition Problem with the Help of Ants
May 21, 2014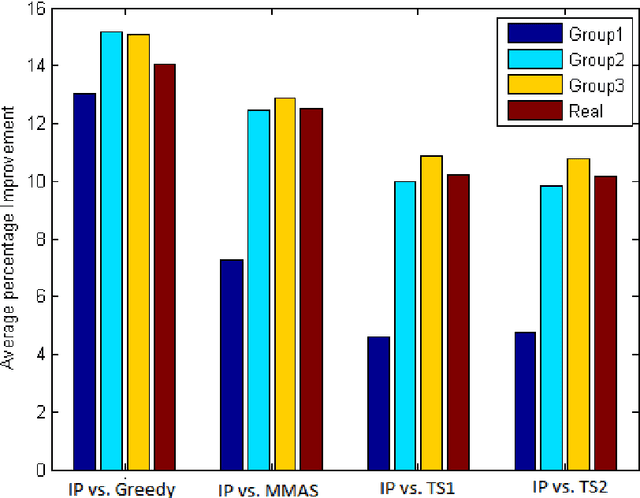
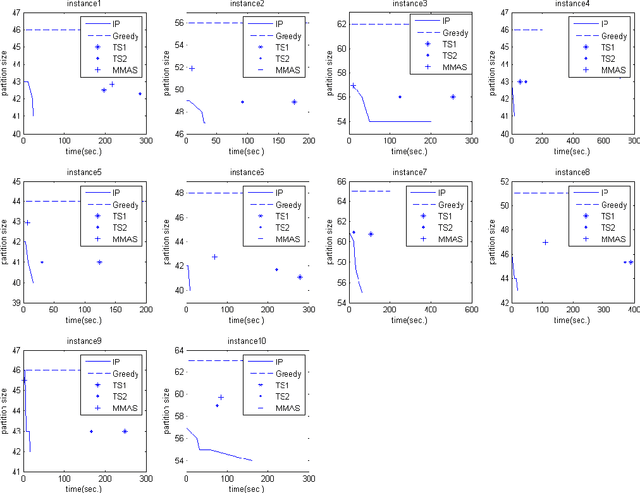
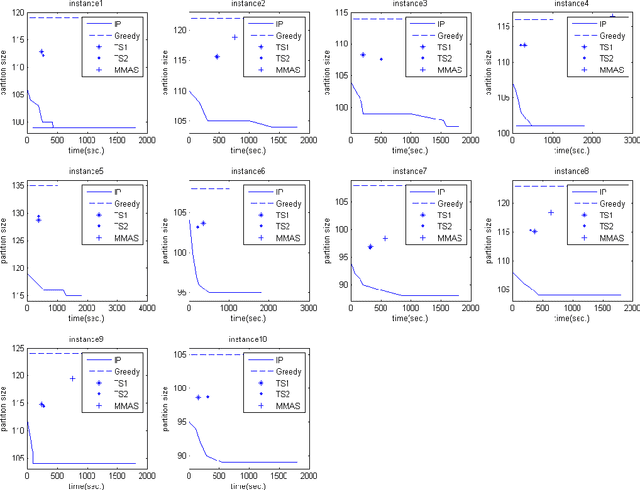
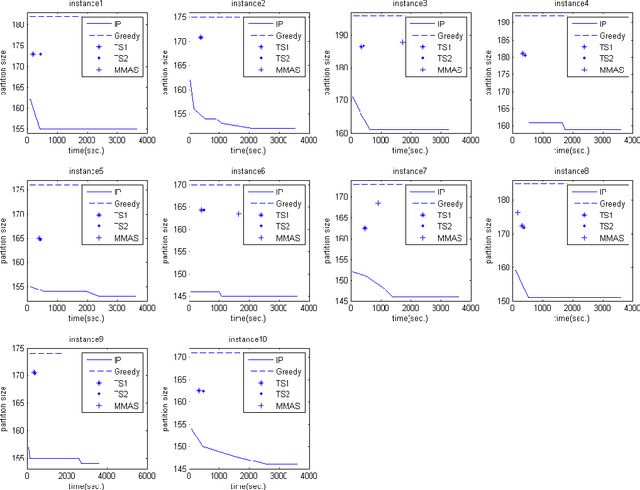
In this paper, we consider the problem of finding a minimum common partition of two strings. The problem has its application in genome comparison. As it is an NP-hard, discrete combinatorial optimization problem, we employ a metaheuristic technique, namely, MAX-MIN ant system to solve this problem. To achieve better efficiency we first map the problem instance into a special kind of graph. Subsequently, we employ a MAX-MIN ant system to achieve high quality solutions for the problem. Experimental results show the superiority of our algorithm in comparison with the state of art algorithm in the literature. The improvement achieved is also justified by standard statistical test.