 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEntropy, Disagreement, and the Limits of Foundation Models in Genomics
Apr 05, 2026Foundation models in genomics have shown mixed success compared to their counterparts in natural language processing. Yet, the reasons for their limited effectiveness remain poorly understood. In this work, we investigate the role of entropy as a fundamental factor limiting the capacities of such models to learn from their training data and develop foundational capabilities. We train ensembles of models on text and DNA sequences and analyze their predictions, static embeddings, and empirical Fisher information flow. We show that the high entropy of genomic sequences -- from the point of view of unseen token prediction -- leads to near-uniform output distributions, disagreement across models, and unstable static embeddings, even for models that are matched in architecture, training and data. We then demonstrate that models trained on DNA concentrate Fisher information in embedding layers, seemingly failing to exploit inter-token relationships. Our results suggest that self-supervised training from sequences alone may not be applicable to genomic data, calling into question the assumptions underlying current methodologies for training genomic foundation models.
Learning to Untangle Genome Assembly with Graph Convolutional Networks
Jun 01, 2022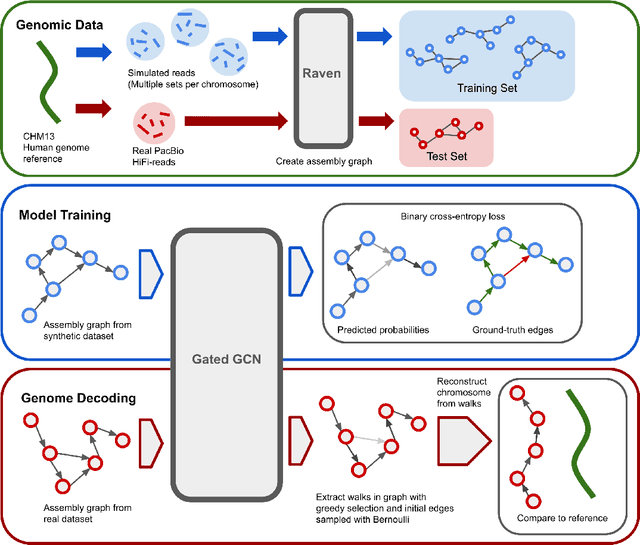
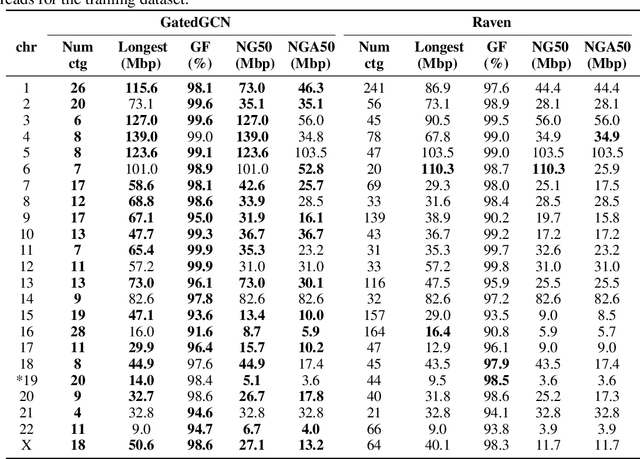
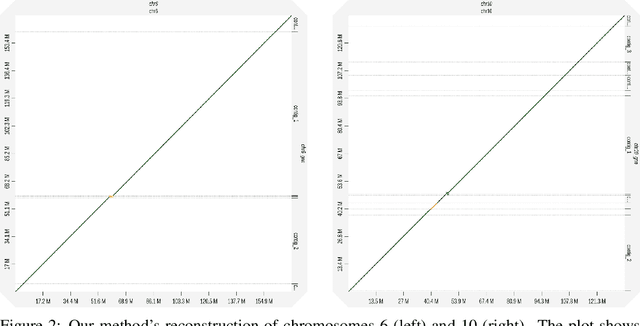
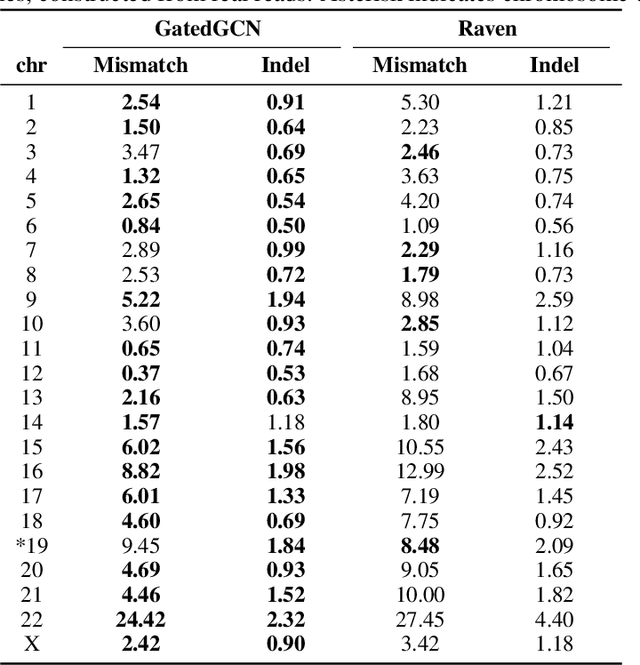
A quest to determine the complete sequence of a human DNA from telomere to telomere started three decades ago and was finally completed in 2021. This accomplishment was a result of a tremendous effort of numerous experts who engineered various tools and performed laborious manual inspection to achieve the first gapless genome sequence. However, such method can hardly be used as a general approach to assemble different genomes, especially when the assembly speed is critical given the large amount of data. In this work, we explore a different approach to the central part of the genome assembly task that consists of untangling a large assembly graph from which a genomic sequence needs to be reconstructed. Our main motivation is to reduce human-engineered heuristics and use deep learning to develop more generalizable reconstruction techniques. Precisely, we introduce a new learning framework to train a graph convolutional network to resolve assembly graphs by finding a correct path through them. The training is supervised with a dataset generated from the resolved CHM13 human sequence and tested on assembly graphs built using real human PacBio HiFi reads. Experimental results show that a model, trained on simulated graphs generated solely from a single chromosome, is able to remarkably resolve all other chromosomes. Moreover, the model outperforms hand-crafted heuristics from a state-of-the-art \textit{de novo} assembler on the same graphs. Reconstructed chromosomes with graph networks are more accurate on nucleotide level, report lower number of contigs, higher genome reconstructed fraction and NG50/NGA50 assessment metrics.
A step towards neural genome assembly
Nov 10, 2020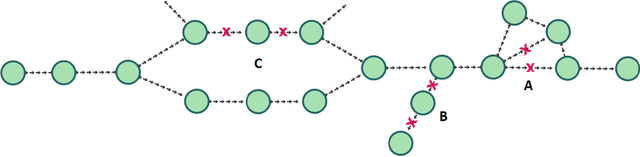
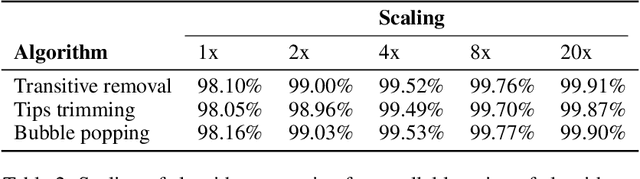
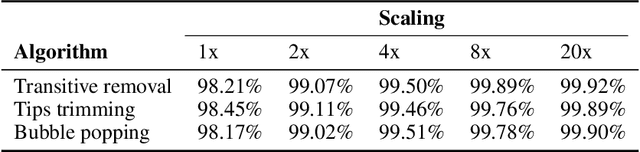

De novo genome assembly focuses on finding connections between a vast amount of short sequences in order to reconstruct the original genome. The central problem of genome assembly could be described as finding a Hamiltonian path through a large directed graph with a constraint that an unknown number of nodes and edges should be avoided. However, due to local structures in the graph and biological features, the problem can be reduced to graph simplification, which includes removal of redundant information. Motivated by recent advancements in graph representation learning and neural execution of algorithms, in this work we train the MPNN model with max-aggregator to execute several algorithms for graph simplification. We show that the algorithms were learned successfully and can be scaled to graphs of sizes up to 20 times larger than the ones used in training. We also test on graphs obtained from real-world genomic data---that of a lambda phage and E. coli.