 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRegression Transformer: Concurrent Conditional Generation and Regression by Blending Numerical and Textual Tokens
Feb 01, 2022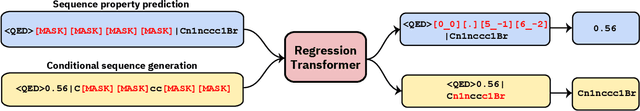
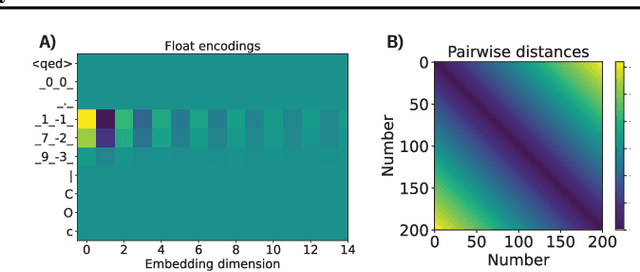
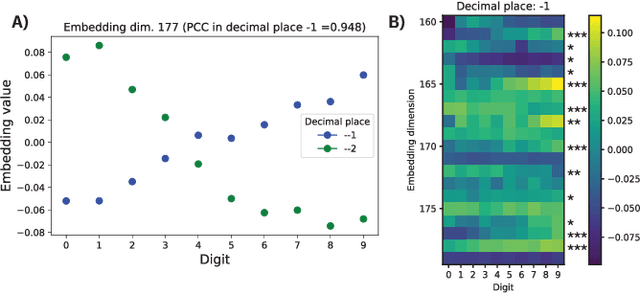
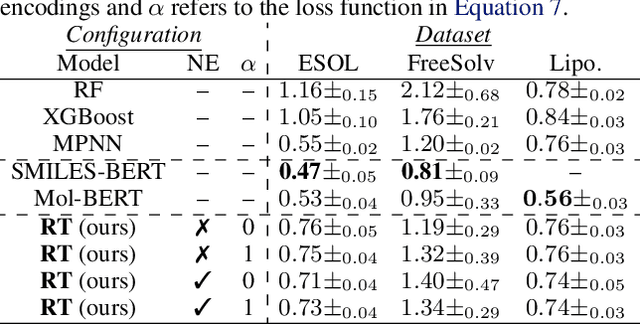
We report the Regression Transformer (RT), a method that abstracts regression as a conditional sequence modeling problem. The RT casts continuous properties as sequences of numerical tokens and encodes them jointly with conventional tokens. This yields a dichotomous model that can seamlessly transition between solving regression tasks and conditional generation tasks; solely governed by the mask location. We propose several extensions to the XLNet objective and adopt an alternating training scheme to concurrently optimize property prediction and conditional text generation based on a self-consistency loss. Our experiments on both chemical and protein languages demonstrate that the performance of traditional regression models can be surpassed despite training with cross entropy loss. Importantly, priming the same model with continuous properties yields a highly competitive conditional generative models that outperforms specialized approaches in a constrained property optimization benchmark. In sum, the Regression Transformer opens the door for "swiss army knife" models that excel at both regression and conditional generation. This finds application particularly in property-driven, local exploration of the chemical or protein space.
TITAN: T Cell Receptor Specificity Prediction with Bimodal Attention Networks
Apr 21, 2021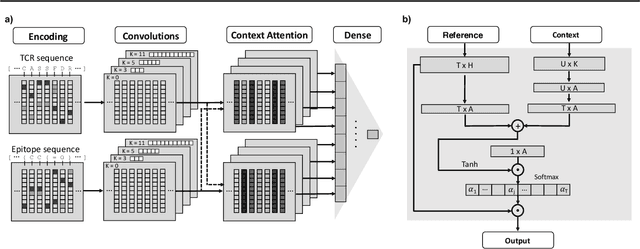

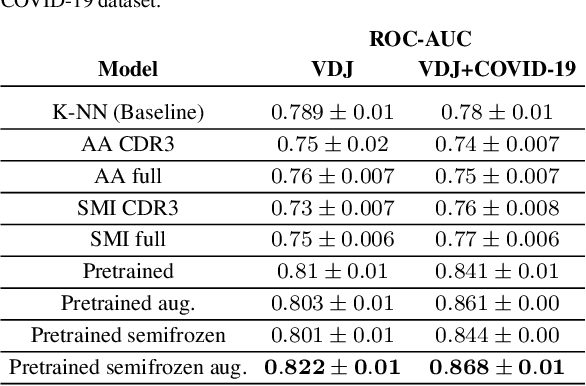
Motivation: The activity of the adaptive immune system is governed by T-cells and their specific T-cell receptors (TCR), which selectively recognize foreign antigens. Recent advances in experimental techniques have enabled sequencing of TCRs and their antigenic targets (epitopes), allowing to research the missing link between TCR sequence and epitope binding specificity. Scarcity of data and a large sequence space make this task challenging, and to date only models limited to a small set of epitopes have achieved good performance. Here, we establish a k-nearest-neighbor (K-NN) classifier as a strong baseline and then propose TITAN (Tcr epITope bimodal Attention Networks), a bimodal neural network that explicitly encodes both TCR sequences and epitopes to enable the independent study of generalization capabilities to unseen TCRs and/or epitopes. Results: By encoding epitopes at the atomic level with SMILES sequences, we leverage transfer learning and data augmentation to enrich the input data space and boost performance. TITAN achieves high performance in the prediction of specificity of unseen TCRs (ROC-AUC 0.87 in 10-fold CV) and surpasses the results of the current state-of-the-art (ImRex) by a large margin. Notably, our Levenshtein-distance-based K-NN classifier also exhibits competitive performance on unseen TCRs. While the generalization to unseen epitopes remains challenging, we report two major breakthroughs. First, by dissecting the attention heatmaps, we demonstrate that the sparsity of available epitope data favors an implicit treatment of epitopes as classes. This may be a general problem that limits unseen epitope performance for sufficiently complex models. Second, we show that TITAN nevertheless exhibits significantly improved performance on unseen epitopes and is capable of focusing attention on chemically meaningful molecular structures.
Accelerating COVID-19 Differential Diagnosis with Explainable Ultrasound Image Analysis
Sep 13, 2020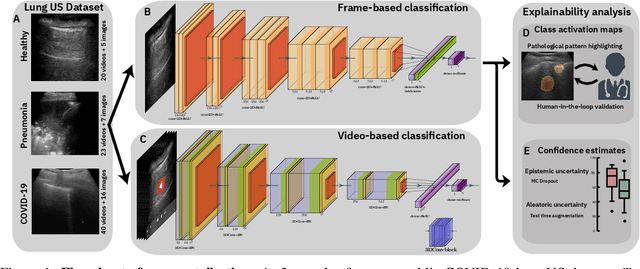

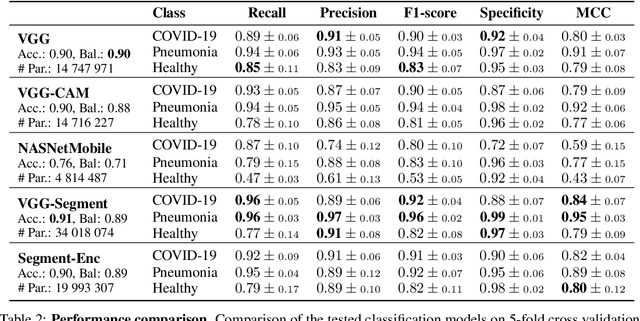
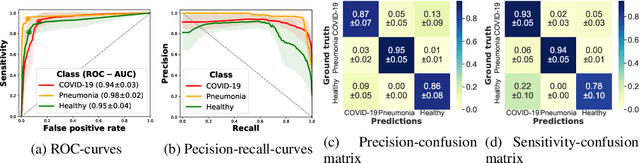
Controlling the COVID-19 pandemic largely hinges upon the existence of fast, safe, and highly-available diagnostic tools. Ultrasound, in contrast to CT or X-Ray, has many practical advantages and can serve as a globally-applicable first-line examination technique. We provide the largest publicly available lung ultrasound (US) dataset for COVID-19 consisting of 106 videos from three classes (COVID-19, bacterial pneumonia, and healthy controls); curated and approved by medical experts. On this dataset, we perform an in-depth study of the value of deep learning methods for differential diagnosis of COVID-19. We propose a frame-based convolutional neural network that correctly classifies COVID-19 US videos with a sensitivity of 0.98+-0.04 and a specificity of 0.91+-08 (frame-based sensitivity 0.93+-0.05, specificity 0.87+-0.07). We further employ class activation maps for the spatio-temporal localization of pulmonary biomarkers, which we subsequently validate for human-in-the-loop scenarios in a blindfolded study with medical experts. Aiming for scalability and robustness, we perform ablation studies comparing mobile-friendly, frame- and video-based architectures and show reliability of the best model by aleatoric and epistemic uncertainty estimates. We hope to pave the road for a community effort toward an accessible, efficient and interpretable screening method and we have started to work on a clinical validation of the proposed method. Data and code are publicly available.
PaccMann$^{RL}$ on SARS-CoV-2: Designing antiviral candidates with conditional generative models
May 31, 2020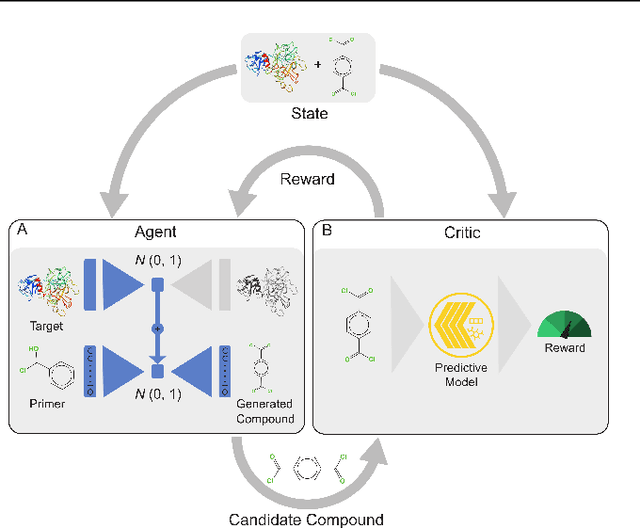

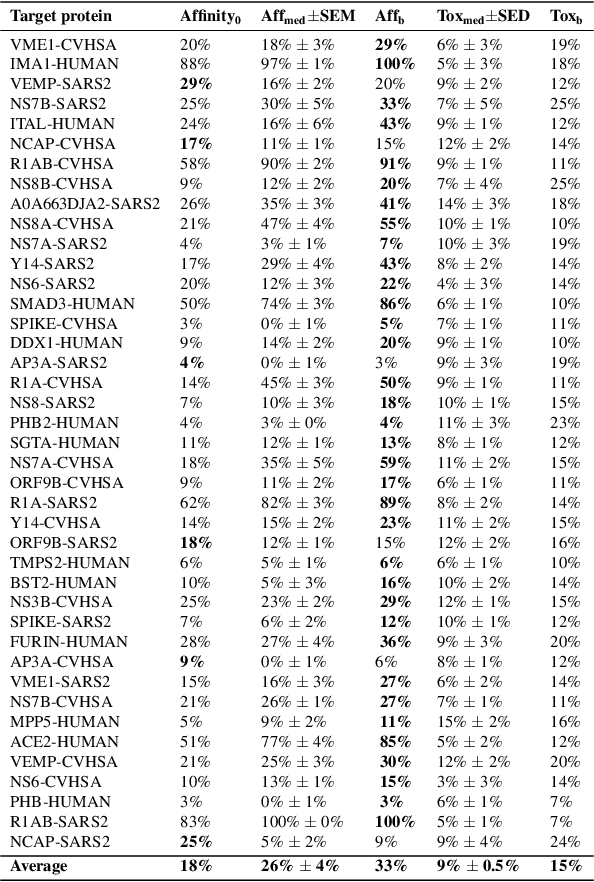
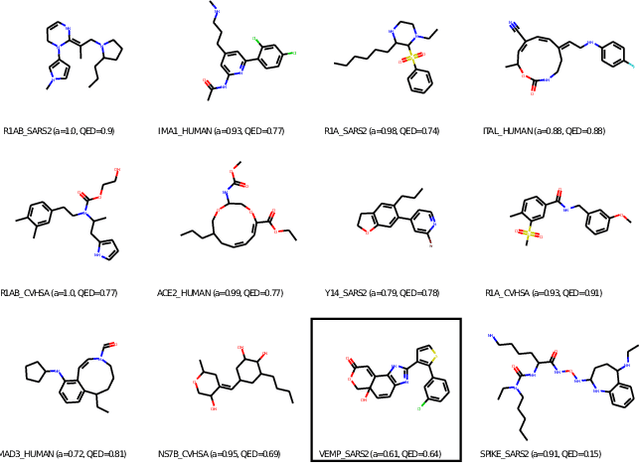
With the fast development of COVID-19 into a global pandemic, scientists around the globe are desperately searching for effective antiviral therapeutic agents. Bridging systems biology and drug discovery, we propose a deep learning framework for conditional de novo design of antiviral candidate drugs tailored against given protein targets. First, we train a multimodal ligand--protein binding affinity model on predicting affinities of antiviral compounds to target proteins and couple this model with pharmacological toxicity predictors. Exploiting this multi-objective as a reward function of a conditional molecular generator (consisting of two VAEs), we showcase a framework that navigates the chemical space toward regions with more antiviral molecules. Specifically, we explore a challenging setting of generating ligands against unseen protein targets by performing a leave-one-out-cross-validation on 41 SARS-CoV-2-related target proteins. Using deep RL, it is demonstrated that in 35 out of 41 cases, the generation is biased towards sampling more binding ligands, with an average increase of 83% comparing to an unbiased VAE. We present a case-study on a potential Envelope-protein inhibitor and perform a synthetic accessibility assessment of the best generated molecules is performed that resembles a viable roadmap towards a rapid in-vitro evaluation of potential SARS-CoV-2 inhibitors.
POCOVID-Net: Automatic Detection of COVID-19 From a New Lung Ultrasound Imaging Dataset
May 05, 2020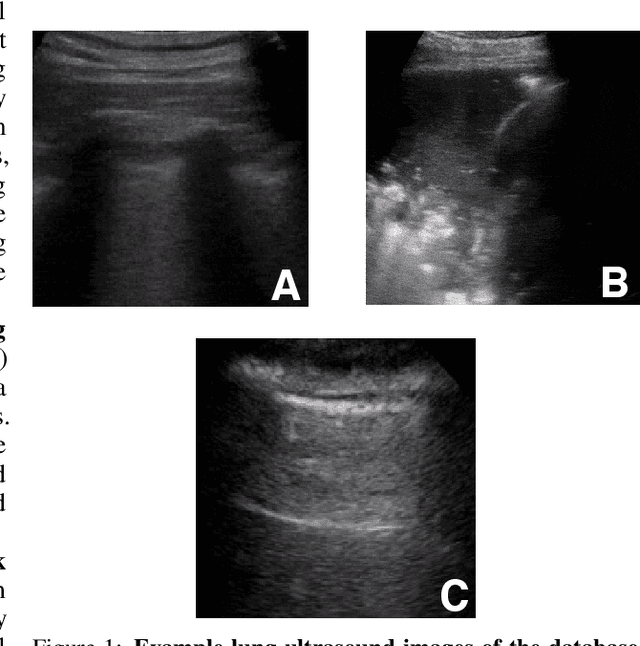
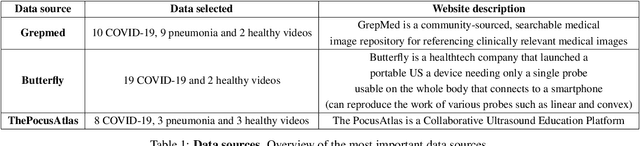
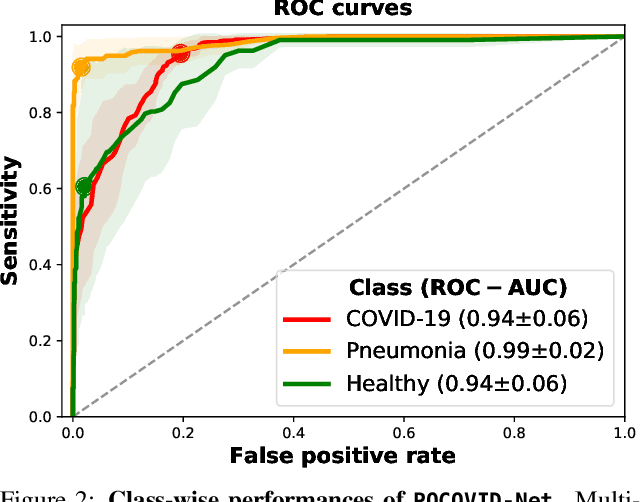
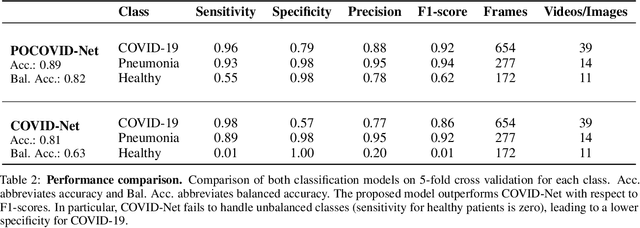
With the rapid development of COVID-19 into a global pandemic, there is an ever more urgent need for cheap, fast and reliable tools that can assist physicians in diagnosing COVID-19. Medical imaging such as CT can take a key role in complementing conventional diagnostic tools from molecular biology, and, using deep learning techniques, several automatic systems were demonstrated promising performances using CT or X-ray data. Here, we advocate a more prominent role of point-of-care ultrasound imaging to guide COVID-19 detection. Ultrasound is non-invasive and ubiquitous in medical facilities around the globe. Our contribution is threefold. First, we gather a lung ultrasound (POCUS) dataset consisting of (currently) 1103 images (654 COVID-19, 277 bacterial pneumonia and 172 healthy controls), sampled from 64 videos. While this dataset was assembled from various online sources and is by no means exhaustive, it was processed specifically to feed deep learning models and is intended to serve as a starting point for an open-access initiative. Second, we train a deep convolutional neural network (POCOVID-Net) on this 3-class dataset and achieve an accuracy of 89% and, by a majority vote, a video accuracy of 92% . For detecting COVID-19 in particular, the model performs with a sensitivity of 0.96, a specificity of 0.79 and F1-score of 0.92 in a 5-fold cross validation. Third, we provide an open-access web service (POCOVIDScreen) that is available at: https://pocovidscreen.org. The website deploys the predictive model, allowing to perform predictions on ultrasound lung images. In addition, it grants medical staff the option to (bulk) upload their own screenings in order to contribute to the growing public database of pathological lung ultrasound images. Dataset and code are available from: https://github.com/jannisborn/covid19_pocus_ultrasound
Reinforcement learning-driven de-novo design of anticancer compounds conditioned on biomolecular profiles
Aug 29, 2019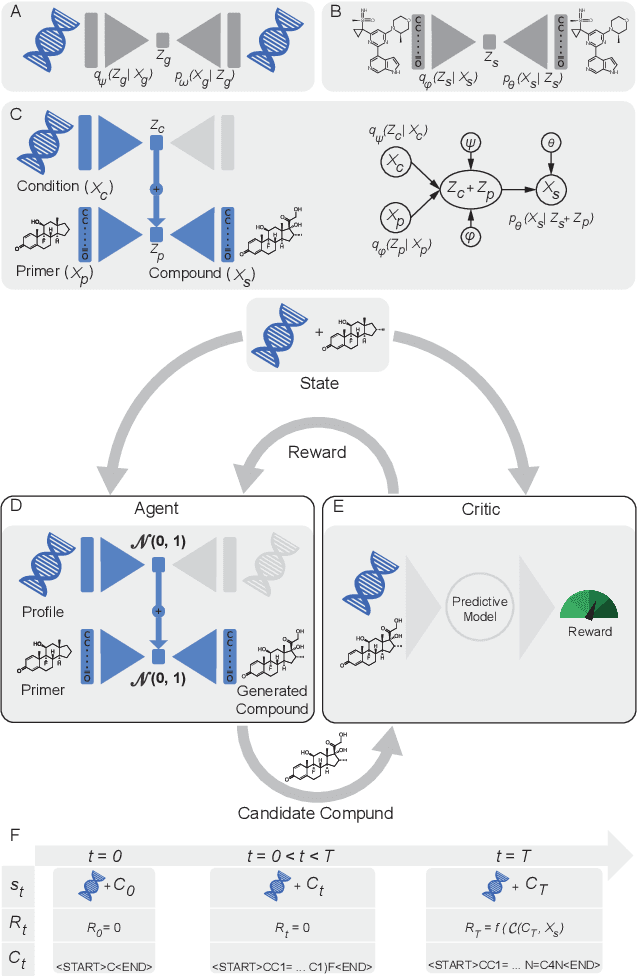
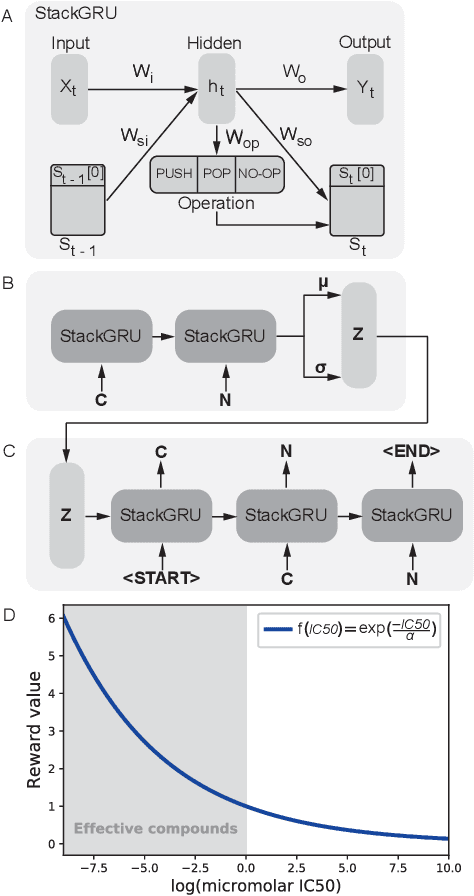
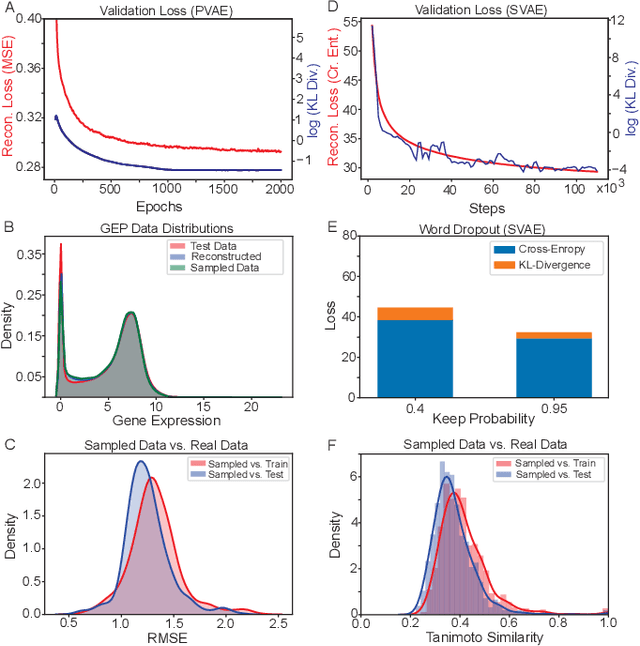
With the advent of deep generative models in computational chemistry, in silico anticancer drug design has undergone an unprecedented transformation. While state-of-the-art deep learning approaches have shown potential in generating compounds with desired chemical properties, they entirely overlook the genetic profile and properties of the target disease. In the case of cancer, this is problematic since it is a highly genetic disease in which the biomolecular profile of target cells determines the response to therapy. Here, we introduce the first deep generative model capable of generating anticancer compounds given a target biomolecular profile. Using a reinforcement learning framework, the transcriptomic profile of cancer cells is used as a context in which anticancer molecules are generated and optimized to obtain effective compounds for the given profile. Our molecule generator combines two pretrained variational autoencoders (VAEs) and a multimodal efficacy predictor - the first VAE generates transcriptomic profiles while the second conditional VAE generates novel molecular structures conditioned on the given transcriptomic profile. The efficacy predictor is used to optimize the generated molecules through a reward determined by the predicted IC50 drug sensitivity for the generated molecule and the target profile. We demonstrate how the molecule generation can be biased towards compounds with high inhibitory effect against individual cell lines or specific cancer sites. We verify our approach by investigating candidate drugs generated against specific cancer types and investigate their structural similarity to existing compounds with known efficacy against these cancer types. We envision our approach to transform in silico anticancer drug design by increasing success rates in lead compound discovery via leveraging the biomolecular characteristics of the disease.
Towards Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-based Convolutional Encoders
May 22, 2019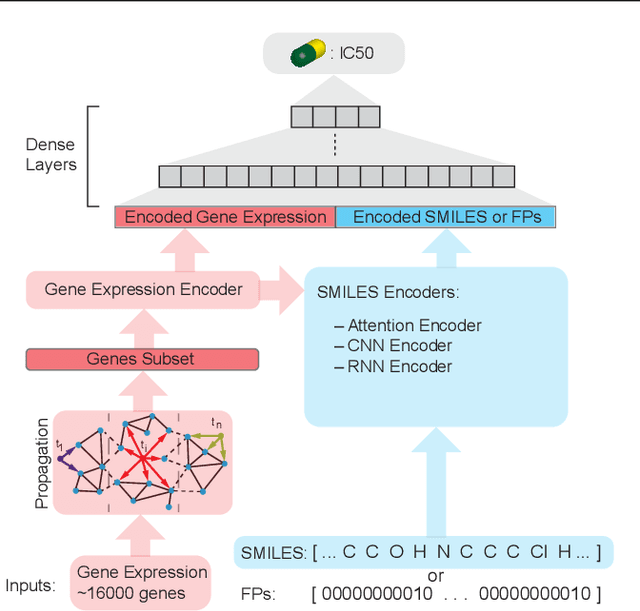
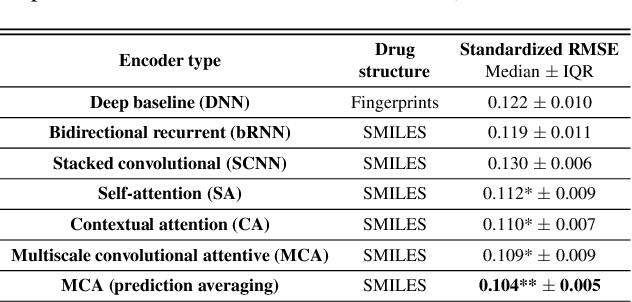
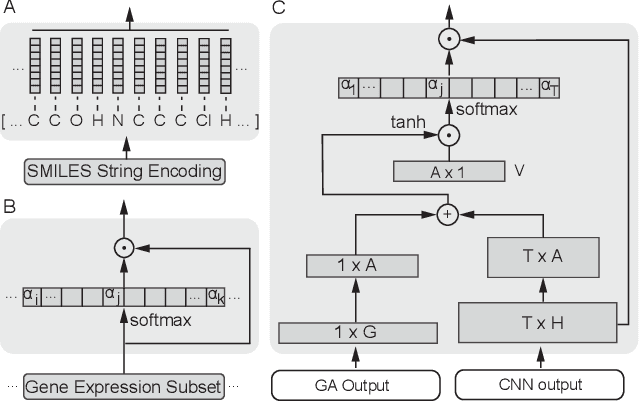
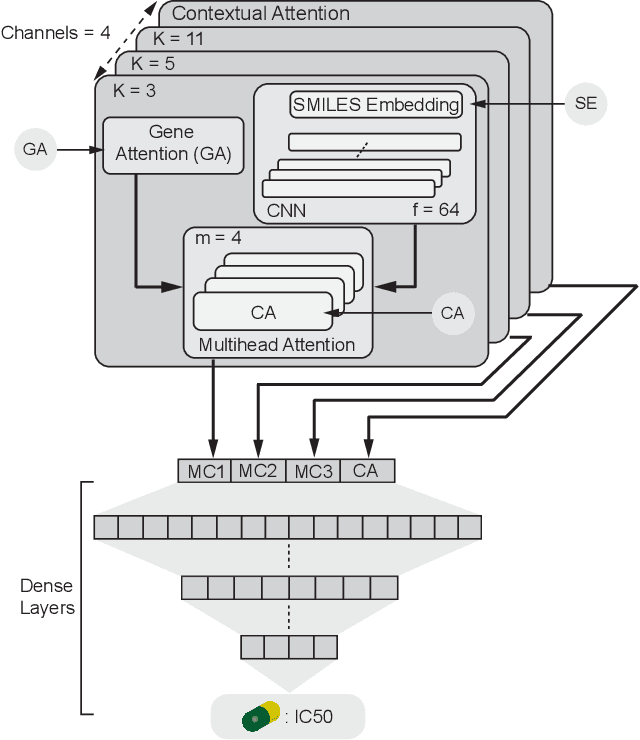
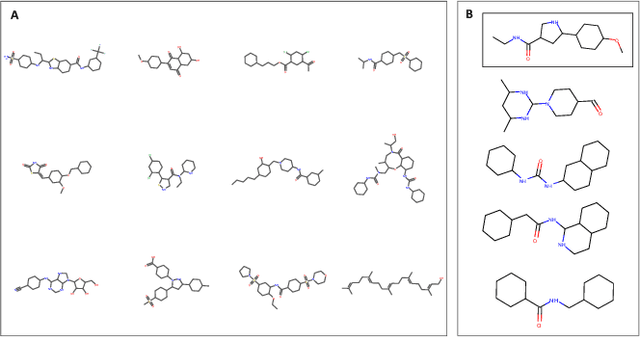
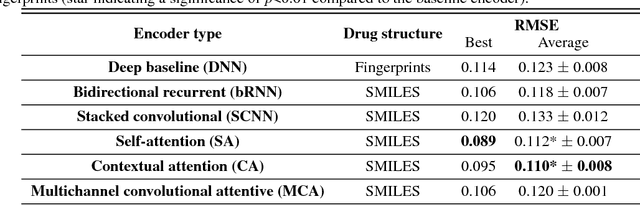
In line with recent advances in neural drug design and sensitivity prediction, we propose a novel architecture for interpretable prediction of anticancer compound sensitivity using a multimodal attention-based convolutional encoder. Our model is based on the three key pillars of drug sensitivity: compounds' structure in the form of a SMILES sequence, gene expression profiles of tumors and prior knowledge on intracellular interactions from protein-protein interaction networks. We demonstrate that our multiscale convolutional attention-based (MCA) encoder significantly outperforms a baseline model trained on Morgan fingerprints, a selection of encoders based on SMILES as well as previously reported state of the art for multimodal drug sensitivity prediction (R2 = 0.86 and RMSE = 0.89). Moreover, the explainability of our approach is demonstrated by a thorough analysis of the attention weights. We show that the attended genes significantly enrich apoptotic processes and that the drug attention is strongly correlated with a standard chemical structure similarity index. Finally, we report a case study of two receptor tyrosine kinase (RTK) inhibitors acting on a leukemia cell line, showcasing the ability of the model to focus on informative genes and submolecular regions of the two compounds. The demonstrated generalizability and the interpretability of our model testify its potential for in-silico prediction of anticancer compound efficacy on unseen cancer cells, positioning it as a valid solution for the development of personalized therapies as well as for the evaluation of candidate compounds in de novo drug design.
PaccMann: Prediction of anticancer compound sensitivity with multi-modal attention-based neural networks
Nov 16, 2018
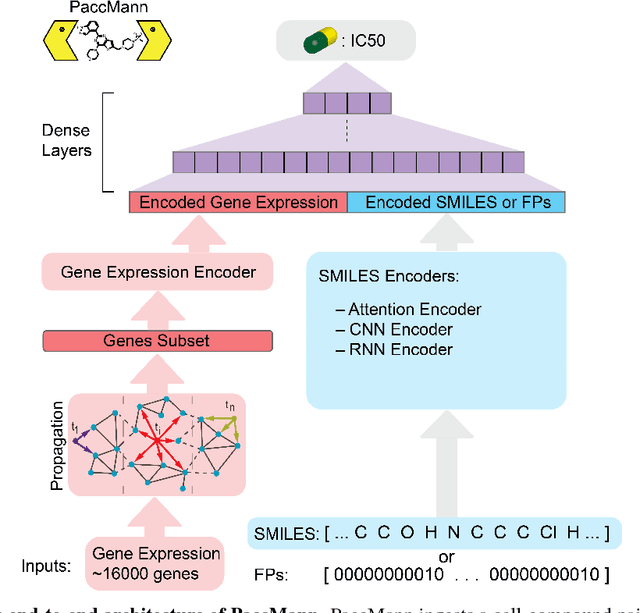
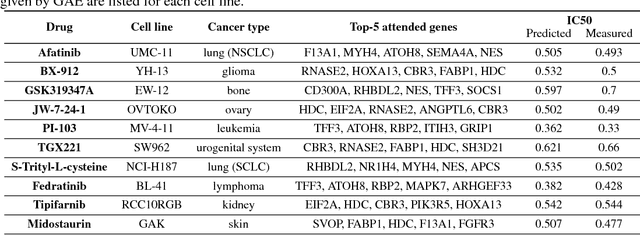
We present a novel approach for the prediction of anticancer compound sensitivity by means of multi-modal attention-based neural networks (PaccMann). In our approach, we integrate three key pillars of drug sensitivity, namely, the molecular structure of compounds, transcriptomic profiles of cancer cells as well as prior knowledge about interactions among proteins within cells. Our models ingest a drug-cell pair consisting of SMILES encoding of a compound and the gene expression profile of a cancer cell and predicts an IC50 sensitivity value. Gene expression profiles are encoded using an attention-based encoding mechanism that assigns high weights to the most informative genes. We present and study three encoders for SMILES string of compounds: 1) bidirectional recurrent 2) convolutional 3) attention-based encoders. We compare our devised models against a baseline model that ingests engineered fingerprints to represent the molecular structure. We demonstrate that using our attention-based encoders, we can surpass the baseline model. The use of attention-based encoders enhance interpretability and enable us to identify genes, bonds and atoms that were used by the network to make a prediction.