 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking Time Series Forecasting Models: From Statistical Techniques to Foundation Models in Real-World Applications
Feb 05, 2025



Time series forecasting is essential for operational intelligence in the hospitality industry, and particularly challenging in large-scale, distributed systems. This study evaluates the performance of statistical, machine learning (ML), deep learning, and foundation models in forecasting hourly sales over a 14-day horizon using real-world data from a network of thousands of restaurants across Germany. The forecasting solution includes features such as weather conditions, calendar events, and time-of-day patterns. Results demonstrate the strong performance of ML-based meta-models and highlight the emerging potential of foundation models like Chronos and TimesFM, which deliver competitive performance with minimal feature engineering, leveraging only the pre-trained model (zero-shot inference). Additionally, a hybrid PySpark-Pandas approach proves to be a robust solution for achieving horizontal scalability in large-scale deployments.
PEvoLM: Protein Sequence Evolutionary Information Language Model
Aug 16, 2023With the exponential increase of the protein sequence databases over time, multiple-sequence alignment (MSA) methods, like PSI-BLAST, perform exhaustive and time-consuming database search to retrieve evolutionary information. The resulting position-specific scoring matrices (PSSMs) of such search engines represent a crucial input to many machine learning (ML) models in the field of bioinformatics and computational biology. A protein sequence is a collection of contiguous tokens or characters called amino acids (AAs). The analogy to natural language allowed us to exploit the recent advancements in the field of Natural Language Processing (NLP) and therefore transfer NLP state-of-the-art algorithms to bioinformatics. This research presents an Embedding Language Model (ELMo), converting a protein sequence to a numerical vector representation. While the original ELMo trained a 2-layer bidirectional Long Short-Term Memory (LSTMs) network following a two-path architecture, one for the forward and the second for the backward pass, by merging the idea of PSSMs with the concept of transfer-learning, this work introduces a novel bidirectional language model (bi-LM) with four times less free parameters and using rather a single path for both passes. The model was trained not only on predicting the next AA but also on the probability distribution of the next AA derived from similar, yet different sequences as summarized in a PSSM, simultaneously for multi-task learning, hence learning evolutionary information of protein sequences as well. The network architecture and the pre-trained model are made available as open source under the permissive MIT license on GitHub at https://github.com/issararab/PEvoLM.
ToxTree: descriptor-based machine learning models for both hERG and Nav1.5 cardiotoxicity liability predictions
Dec 27, 2021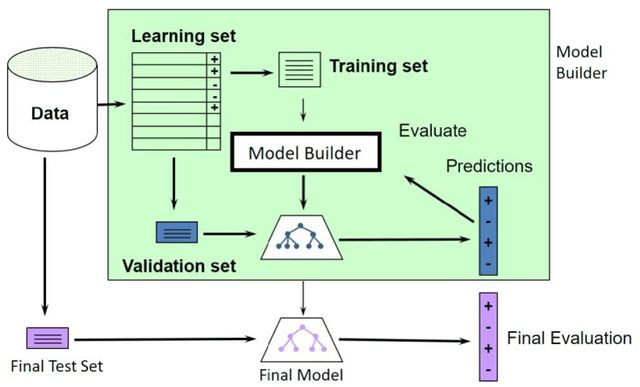
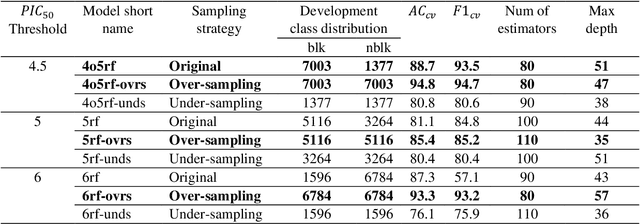
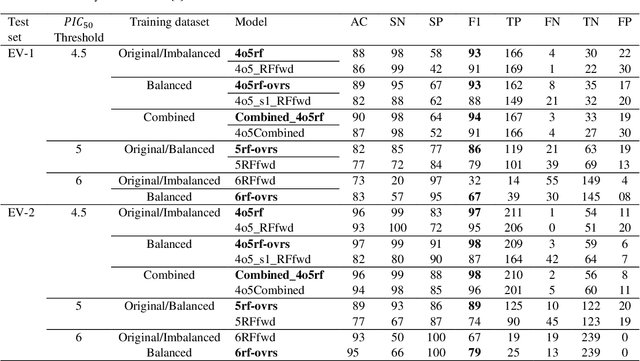
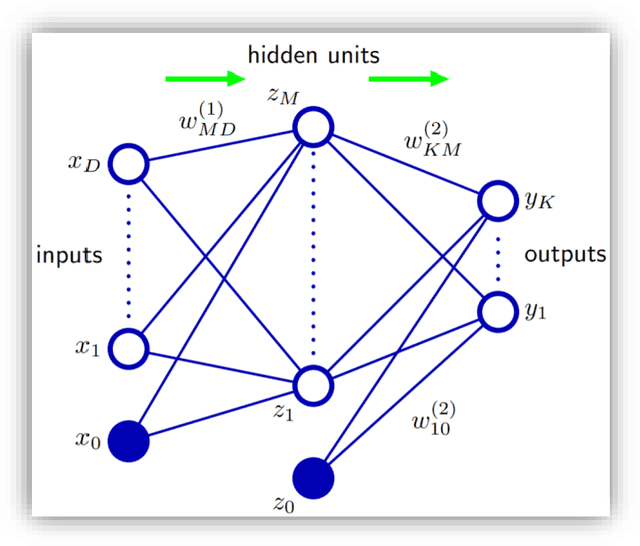
Drug-mediated blockade of the voltage-gated potassium channel(hERG) and the voltage-gated sodium channel (Nav1.5) can lead to severe cardiovascular complications. This rising concern has been reflected in the drug development arena, as the frequent emergence of cardiotoxicity from many approved drugs led to either discontinuing their use or, in some cases, their withdrawal from the market. Predicting potential hERG and Nav1.5 blockers at the outset of the drug discovery process can resolve this problem and can, therefore, decrease the time and expensive cost of developing safe drugs. One fast and cost-effective approach is to use in silico predictive methods to weed out potential hERG and Nav1.5 blockers at the early stages of drug development. Here, we introduce two robust 2D descriptor-based QSAR predictive models for both hERG and Nav1.5 liability predictions. The machine learning models were trained for both regression, predicting the potency value of a drug, and multiclass classification at three different potency cut-offs (i.e. 1$\mu$M, 10$\mu$M, and 30$\mu$M), where ToxTree-hERG Classifier, a pipeline of Random Forest models, was trained on a large curated dataset of 8380 unique molecular compounds. Whereas ToxTree-Nav1.5 Classifier, a pipeline of kernelized SVM models, was trained on a large manually curated set of 1550 unique compounds retrieved from both ChEMBL and PubChem publicly available bioactivity databases. The proposed hERG inducer outperformed most metrics of the state-of-the-art published model and other existing tools. Additionally, we are introducing the first Nav1.5 liability predictive model achieving a Q4 = 74.9% and a binary classification of Q2 = 86.7% with MCC = 71.2% evaluated on an external test set of 173 unique compounds. The curated datasets used in this project are made publicly available to the research community.