 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Phase Prediction to Phase Design: A ReAct Agent Framework for High-Entropy Alloy Discovery
Mar 10, 2026Discovering high-entropy alloy (HEA) compositions that reliably form a target crystal phase is a high-dimensional inverse design problem that conventional trial-and-error experimentation and forward-only machine learning models cannot efficiently solve. Here we present a ReAct (Reasoning + Acting) LLM agent that autonomously proposes, validates, and iteratively refines HEA compositions by querying a calibrated XGBoost surrogate trained on 4,753 experimental records across four phases (FCC, BCC, BCC+FCC, BCC+IM), achieving 94.66\% accuracy (F1 macro = 0.896). Against Bayesian optimisation (BO) and random search baselines, the full-prompt agent achieves descriptor-space rediscovery rates of 38\%, 18\%, and 38\% for FCC, BCC, and BCC+FCC (Mann--Whitney $p \leq 0.039$), with proposals lying 2.4--22.8$\times$ closer to the experimental phase manifold than random search. An ablation reveals that domain priors shift the agent from landmark-alloy recall toward compositionally diverse exploration -- an uninformed agent scores higher rediscovery by concentrating on literature-dense families, while the full-prompt agent explores underrepresented space (unique ratio 1.0 vs.\ 0.39 for BCC+FCC). These regimes represent distinct criteria: proximity to known literature versus genuine discovery. Spearman analysis confirms agent reasoning is statistically aligned with empirical phase distributions ($ρ= 0.736$, $p = 0.004$ for BCC). This work establishes LLM-guided agentic reasoning as a principled, transparent, and manifold-aware complement to gradient-free optimisation for inverse alloy design.
Escaping the Hydrolysis Trap: An Agentic Workflow for Inverse Design of Durable Photocatalytic Covalent Organic Frameworks
Mar 05, 2026Covalent organic frameworks (COFs) are promising photocatalysts for solar hydrogen production, yet the most electronically favorable linkages, imines, hydrolyze rapidly in water, creating a stability--activity trade-off that limits practical deployment. Navigating the combinatorial design space of nodes, linkers, linkages, and functional groups to identify candidates that are simultaneously active and durable remains a formidable challenge. Here we introduce Ara, a large-language-model (LLM) agent that leverages pretrained chemical knowledge, donor--acceptor theory, conjugation effects, and linkage stability hierarchies, to guide the search for photocatalytic COFs satisfying joint band-gap, band-edge, and hydrolytic-stability criteria. Evaluated against random search and Bayesian optimization (BO) over a space consisting of candidates with various nodes, linkers, linkages, and r-groups, screened with a GFN1-xTB fragment pipeline, Ara achieves a 52.7\% hit rate (11.5$\times$ random, p = 0.006), finds its first hit at iteration 12 versus 25 for random search, and significantly outperforms BO (p = 0.006). Inspection of the agent's reasoning traces reveals interpretable chemical logic: early convergence on vinylene and beta-ketoenamine linkages for stability, node selection informed by electron-withdrawing character, and systematic R-group optimization to center the band gap at 2.0 eV. Exhaustive evaluation of the full search space uncovers a complementary exploitation--exploration trade-off between the agent and BO, suggesting that hybrid strategies may combine the strengths of both approaches. These results demonstrate that LLM chemical priors can substantially accelerate multi-criteria materials discovery.
ChemNavigator: Agentic AI Discovery of Design Rules for Organic Photocatalysts
Jan 23, 2026The discovery of high-performance organic photocatalysts for hydrogen evolution remains limited by the vastness of chemical space and the reliance on human intuition for molecular design. Here we present ChemNavigator, an agentic AI system that autonomously derives structure-property relationships through hypothesis-driven exploration of organic photocatalyst candidates. The system integrates large language model reasoning with density functional tight binding calculations in a multi-agent architecture that mirrors the scientific method: formulating hypotheses, designing experiments, executing calculations, and validating findings through rigorous statistical analysis. Through iterative discovery cycles encompassing 200 molecules, ChemNavigator autonomously identified six statistically significant design rules governing frontier orbital energies, including the effects of ether linkages, carbonyl groups, extended conjugation, cyano groups, halogen substituents, and amine groups. Importantly, these rules correspond to established principles of organic electronic structure (resonance donation, inductive withdrawal, $π$-delocalization), demonstrating that the system can independently derive chemical knowledge without explicit programming. Notably, autonomous agentic reasoning extracted these six validated rules from a molecular library where previous ML approaches identified only carbonyl effects. Furthermore, the quantified effect sizes provide a prioritized ranking for synthetic chemists, while feature interaction analysis revealed diminishing returns when combining strategies, challenging additive assumptions in molecular design. This work demonstrates that agentic AI systems can autonomously derive interpretable, chemically grounded design principles, establishing a framework for AI-assisted materials discovery that complements rather than replaces chemical intuition.
Artificial Intelligence in Materials Science and Engineering: Current Landscape, Key Challenges, and Future Trajectorie
Jan 18, 2026Artificial Intelligence is rapidly transforming materials science and engineering, offering powerful tools to navigate complexity, accelerate discovery, and optimize material design in ways previously unattainable. Driven by the accelerating pace of algorithmic advancements and increasing data availability, AI is becoming an essential competency for materials researchers. This review provides a comprehensive and structured overview of the current landscape, synthesizing recent advancements and methodologies for materials scientists seeking to effectively leverage these data-driven techniques. We survey the spectrum of machine learning approaches, from traditional algorithms to advanced deep learning architectures, including CNNs, GNNs, and Transformers, alongside emerging generative AI and probabilistic models such as Gaussian Processes for uncertainty quantification. The review also examines the pivotal role of data in this field, emphasizing how effective representation and featurization strategies, spanning compositional, structural, image-based, and language-inspired approaches, combined with appropriate preprocessing, fundamentally underpin the performance of machine learning models in materials research. Persistent challenges related to data quality, quantity, and standardization, which critically impact model development and application in materials science and engineering, are also addressed.
Teaching Artificial Intelligence to Perform Rapid, Resolution-Invariant Grain Growth Modeling via Fourier Neural Operator
Mar 18, 2025Microstructural evolution, particularly grain growth, plays a critical role in shaping the physical, optical, and electronic properties of materials. Traditional phase-field modeling accurately simulates these phenomena but is computationally intensive, especially for large systems and fine spatial resolutions. While machine learning approaches have been employed to accelerate simulations, they often struggle with resolution dependence and generalization across different grain scales. This study introduces a novel approach utilizing Fourier Neural Operator (FNO) to achieve resolution-invariant modeling of microstructure evolution in multi-grain systems. FNO operates in the Fourier space and can inherently handle varying resolutions by learning mappings between function spaces. By integrating FNO with the phase field method, we developed a surrogate model that significantly reduces computational costs while maintaining high accuracy across different spatial scales. We generated a comprehensive dataset from phase-field simulations using the Fan Chen model, capturing grain evolution over time. Data preparation involved creating input-output pairs with a time shift, allowing the model to predict future microstructures based on current and past states. The FNO-based neural network was trained using sequences of microstructures and demonstrated remarkable accuracy in predicting long-term evolution, even for unseen configurations and higher-resolution grids not encountered during training.
Accelerating phase-field-based simulation via machine learning
May 04, 2022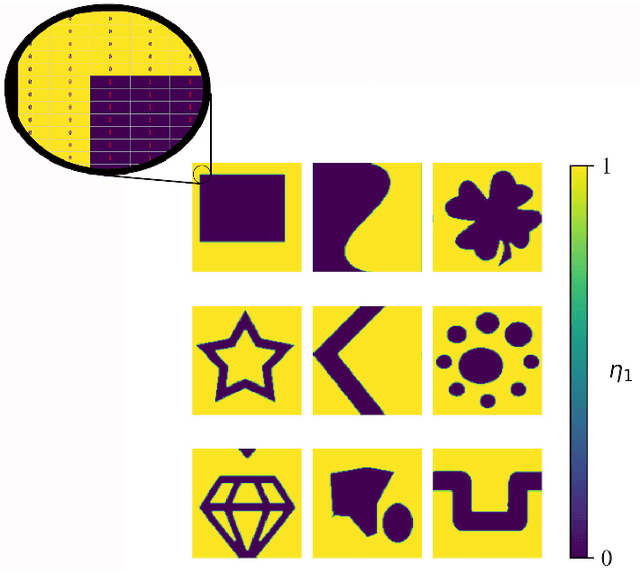
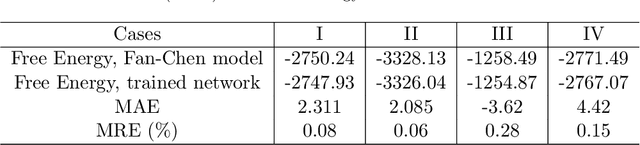
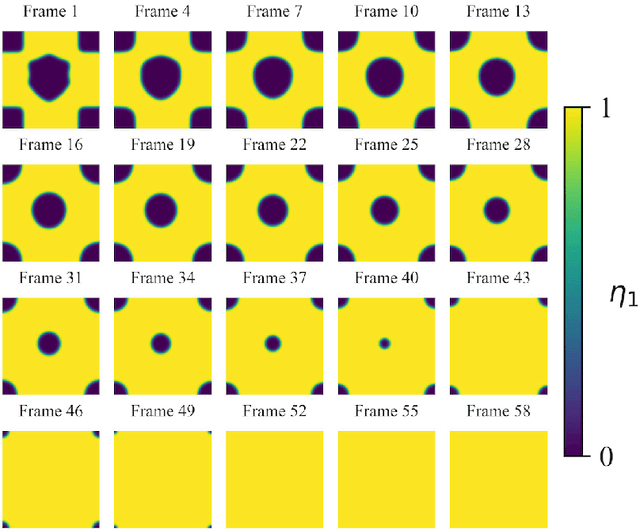
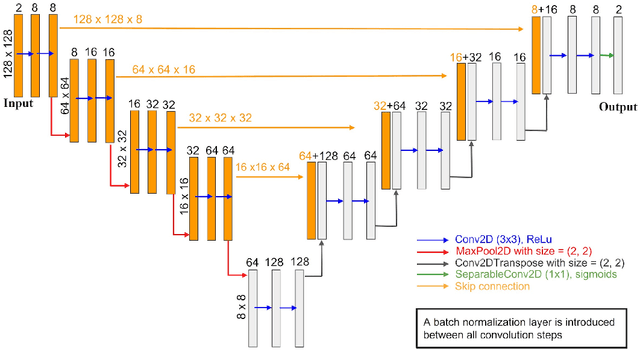
Phase-field-based models have become common in material science, mechanics, physics, biology, chemistry, and engineering for the simulation of microstructure evolution. Yet, they suffer from the drawback of being computationally very costly when applied to large, complex systems. To reduce such computational costs, a Unet-based artificial neural network is developed as a surrogate model in the current work. Training input for this network is obtained from the results of the numerical solution of initial-boundary-value problems (IBVPs) based on the Fan-Chen model for grain microstructure evolution. In particular, about 250 different simulations with varying initial order parameters are carried out and 200 frames of the time evolution of the phase fields are stored for each simulation. The network is trained with 90% of this data, taking the $i$-th frame of a simulation, i.e. order parameter field, as input, and producing the $(i+1)$-th frame as the output. Evaluation of the network is carried out with a test dataset consisting of 2200 microstructures based on different configurations than originally used for training. The trained network is applied recursively on initial order parameters to calculate the time evolution of the phase fields. The results are compared to the ones obtained from the conventional numerical solution in terms of the errors in order parameters and the system's free energy. The resulting order parameter error averaged over all points and all simulation cases is 0.005 and the relative error in the total free energy in all simulation boxes does not exceed 1%.