 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiversity-Aware Reinforcement Learning for de novo Drug Design
Oct 14, 2024



Fine-tuning a pre-trained generative model has demonstrated good performance in generating promising drug molecules. The fine-tuning task is often formulated as a reinforcement learning problem, where previous methods efficiently learn to optimize a reward function to generate potential drug molecules. Nevertheless, in the absence of an adaptive update mechanism for the reward function, the optimization process can become stuck in local optima. The efficacy of the optimal molecule in a local optimization may not translate to usefulness in the subsequent drug optimization process or as a potential standalone clinical candidate. Therefore, it is important to generate a diverse set of promising molecules. Prior work has modified the reward function by penalizing structurally similar molecules, primarily focusing on finding molecules with higher rewards. To date, no study has comprehensively examined how different adaptive update mechanisms for the reward function influence the diversity of generated molecules. In this work, we investigate a wide range of intrinsic motivation methods and strategies to penalize the extrinsic reward, and how they affect the diversity of the set of generated molecules. Our experiments reveal that combining structure- and prediction-based methods generally yields better results in terms of molecular diversity.
Utilizing Reinforcement Learning for de novo Drug Design
Mar 30, 2023Deep learning-based approaches for generating novel drug molecules with specific properties have gained a lot of interest in the last years. Recent studies have demonstrated promising performance for string-based generation of novel molecules utilizing reinforcement learning. In this paper, we develop a unified framework for using reinforcement learning for de novo drug design, wherein we systematically study various on- and off-policy reinforcement learning algorithms and replay buffers to learn an RNN-based policy to generate novel molecules predicted to be active against the dopamine receptor DRD2. Our findings suggest that it is advantageous to use at least both top-scoring and low-scoring molecules for updating the policy when structural diversity is essential. Using all generated molecules at an iteration seems to enhance performance stability for on-policy algorithms. In addition, when replaying high, intermediate, and low-scoring molecules, off-policy algorithms display the potential of improving the structural diversity and number of active molecules generated, but possibly at the cost of a longer exploration phase. Our work provides an open-source framework enabling researchers to investigate various reinforcement learning methods for de novo drug design.
Autonomous Drug Design with Multi-armed Bandits
Jul 04, 2022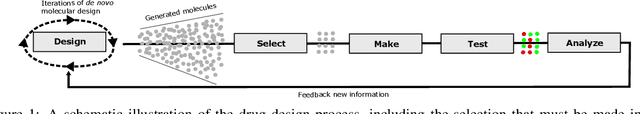
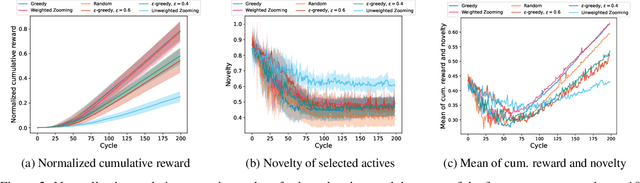
Recent developments in artificial intelligence and automation could potentially enable a new drug design paradigm: autonomous drug design. Under this paradigm, generative models provide suggestions on thousands of molecules with specific properties. However, since only a limited number of molecules can be synthesized and tested, an obvious challenge is how to efficiently select these. We formulate this task as a contextual stochastic multi-armed bandit problem with multiple plays and volatile arms. Then, to solve it, we extend previous work on multi-armed bandits to reflect this setting, and compare our solution with random sampling, greedy selection and decaying-epsilon-greedy selection. To investigate how the different selection strategies affect the cumulative reward and the diversity of the selections, we simulate the drug design process. According to the simulation results, our approach has the potential for better exploring and exploiting the chemical space for autonomous drug design.