 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgeannbatch unlocks terabyte-scale training of biological data in anndata
Apr 02, 2026The scale of biological datasets now routinely exceeds system memory, making data access rather than model computation the primary bottleneck in training machine-learning models. This bottleneck is particularly acute in biology, where widely used community data formats must support heterogeneous metadata, sparse and dense assays, and downstream analysis within established computational ecosystems. Here we present annbatch, a mini-batch loader native to anndata that enables out-of-core training directly on disk-backed datasets. Across single-cell transcriptomics, microscopy and whole-genome sequencing benchmarks, annbatch increases loading throughput by up to an order of magnitude and shortens training from days to hours, while remaining fully compatible with the scverse ecosystem. Annbatch establishes a practical data-loading infrastructure for scalable biological AI, allowing increasingly large and diverse datasets to be used without abandoning standard biological data formats. Github: https://github.com/scverse/annbatch
Conditional out-of-sample generation for unpaired data using trVAE
Oct 30, 2019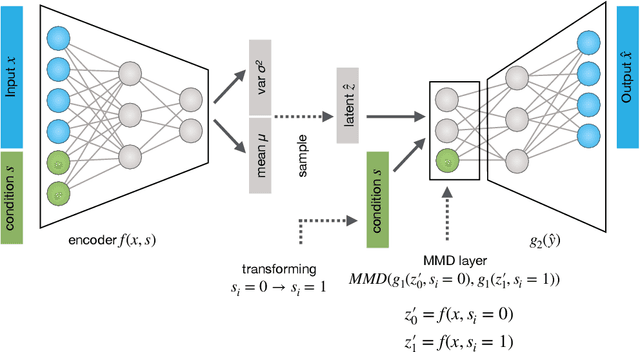
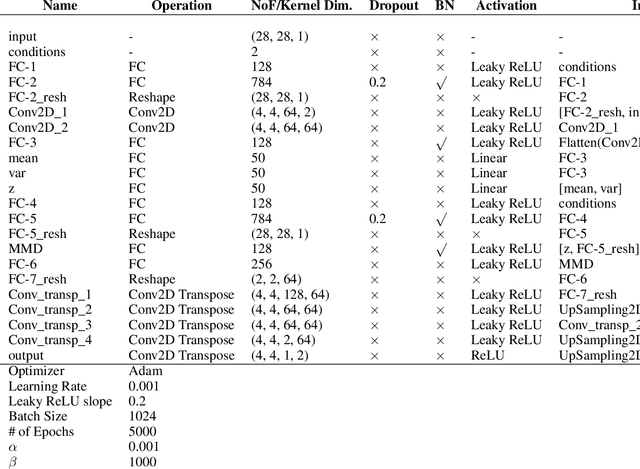
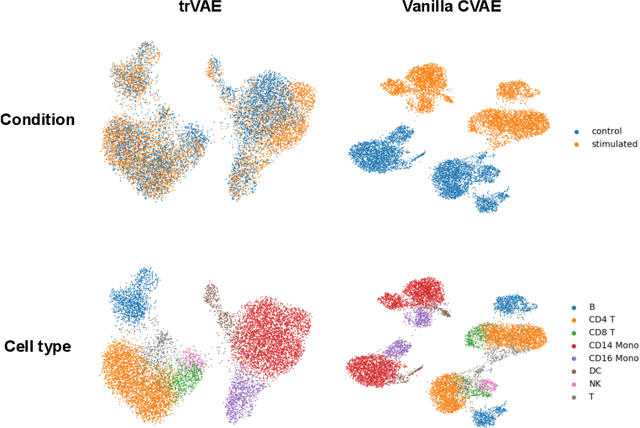
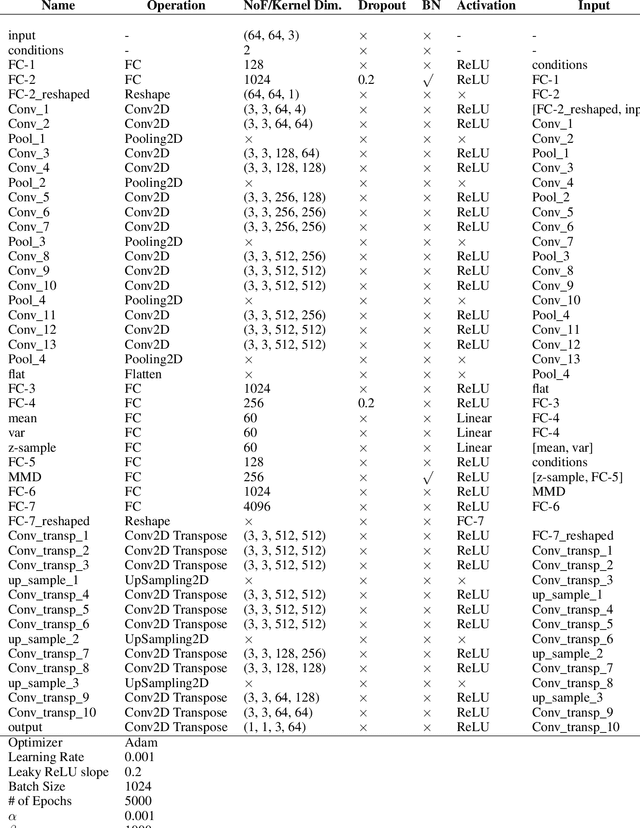
While generative models have shown great success in generating high-dimensional samples conditional on low-dimensional descriptors (learning e.g. stroke thickness in MNIST, hair color in CelebA, or speaker identity in Wavenet), their generation out-of-sample poses fundamental problems. The conditional variational autoencoder (CVAE) as a simple conditional generative model does not explicitly relate conditions during training and, hence, has no incentive of learning a compact joint distribution across conditions. We overcome this limitation by matching their distributions using maximum mean discrepancy (MMD) in the decoder layer that follows the bottleneck. This introduces a strong regularization both for reconstructing samples within the same condition and for transforming samples across conditions, resulting in much improved generalization. We refer to the architecture as \emph{transformer} VAE (trVAE). Benchmarking trVAE on high-dimensional image and tabular data, we demonstrate higher robustness and higher accuracy than existing approaches. In particular, we show qualitatively improved predictions for cellular perturbation response to treatment and disease based on high-dimensional single-cell gene expression data, by tackling previously problematic minority classes and multiple conditions. For generic tasks, we improve Pearson correlations of high-dimensional estimated means and variances with their ground truths from 0.89 to 0.97 and 0.75 to 0.87, respectively.