 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFully transformer-based biomarker prediction from colorectal cancer histology: a large-scale multicentric study
Jan 23, 2023



Background: Deep learning (DL) can extract predictive and prognostic biomarkers from routine pathology slides in colorectal cancer. For example, a DL test for the diagnosis of microsatellite instability (MSI) in CRC has been approved in 2022. Current approaches rely on convolutional neural networks (CNNs). Transformer networks are outperforming CNNs and are replacing them in many applications, but have not been used for biomarker prediction in cancer at a large scale. In addition, most DL approaches have been trained on small patient cohorts, which limits their clinical utility. Methods: In this study, we developed a new fully transformer-based pipeline for end-to-end biomarker prediction from pathology slides. We combine a pre-trained transformer encoder and a transformer network for patch aggregation, capable of yielding single and multi-target prediction at patient level. We train our pipeline on over 9,000 patients from 10 colorectal cancer cohorts. Results: A fully transformer-based approach massively improves the performance, generalizability, data efficiency, and interpretability as compared with current state-of-the-art algorithms. After training on a large multicenter cohort, we achieve a sensitivity of 0.97 with a negative predictive value of 0.99 for MSI prediction on surgical resection specimens. We demonstrate for the first time that resection specimen-only training reaches clinical-grade performance on endoscopic biopsy tissue, solving a long-standing diagnostic problem. Interpretation: A fully transformer-based end-to-end pipeline trained on thousands of pathology slides yields clinical-grade performance for biomarker prediction on surgical resections and biopsies. Our new methods are freely available under an open source license.
Crowdsourcing scoring of immunohistochemistry images: Evaluating Performance of the Crowd and an Automated Computational Method
Jun 23, 2016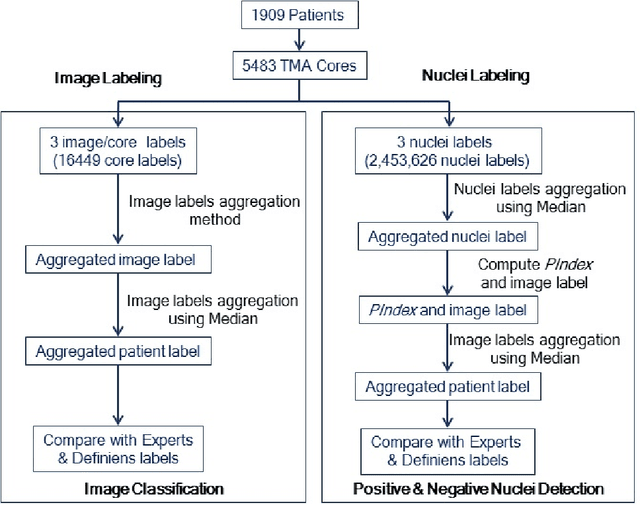
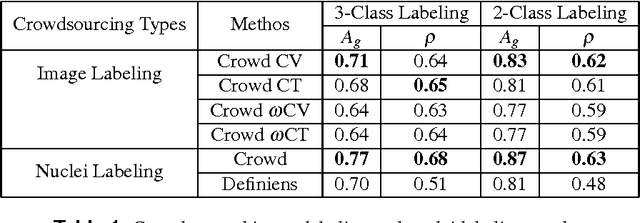
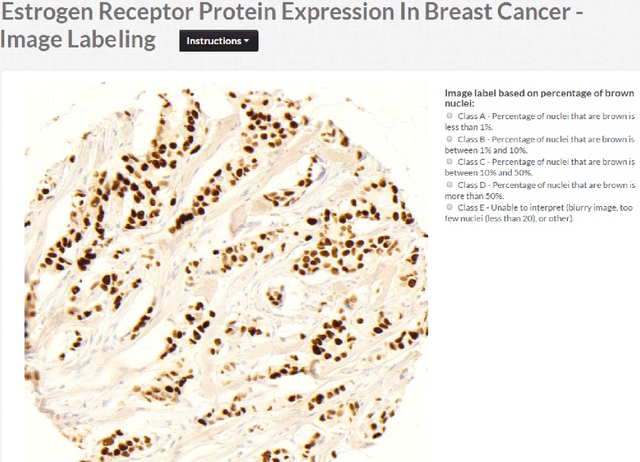
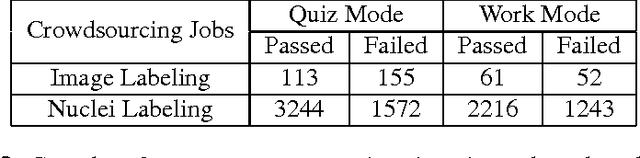
The assessment of protein expression in immunohistochemistry (IHC) images provides important diagnostic, prognostic and predictive information for guiding cancer diagnosis and therapy. Manual scoring of IHC images represents a logistical challenge, as the process is labor intensive and time consuming. Since the last decade, computational methods have been developed to enable the application of quantitative methods for the analysis and interpretation of protein expression in IHC images. These methods have not yet replaced manual scoring for the assessment of IHC in the majority of diagnostic laboratories and in many large-scale research studies. An alternative approach is crowdsourcing the quantification of IHC images to an undefined crowd. The aim of this study is to quantify IHC images for labeling of ER status with two different crowdsourcing approaches, image labeling and nuclei labeling, and compare their performance with automated methods. Crowdsourcing-derived scores obtained greater concordance with the pathologist interpretations for both image labeling and nuclei labeling tasks (83% and 87%), as compared to the pathologist concordance achieved by the automated method (81%) on 5,483 TMA images from 1,909 breast cancer patients. This analysis shows that crowdsourcing the scoring of protein expression in IHC images is a promising new approach for large scale cancer molecular pathology studies.