 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCERN: Correcting Errors in Raw Nanopore Signals Using Hidden Markov Models
Mar 20, 2026Nanopore sequencing can read substantially longer sequences of nucleic acid molecules than other sequencing methods, which has led to advances in genomic analysis such as the gapless human genome assembly. By analyzing the raw electrical signal reads that nanopore sequencing generates from molecules, existing works can map these reads without translating them into DNA characters (i.e., basecalling), allowing for quick and efficient analysis of sequencing data. However, raw signals often contain errors due to noise and mistakes when processing them, which limits the overall accuracy of raw signal analysis. Our goal in this work is to detect and correct errors in raw signals to improve the accuracy of raw signal analyses. To this end, we propose CERN, a mechanism that trains and utilizes a Hidden Markov Model (HMM) to accurately correct signal errors. Our extensive evaluation on various datasets including E. coli, Fruit Fly, and Human genomes shows that CERN 1) consistently improves the overall mapping accuracy of the underlying raw signal analysis tools, 2) minimizes the burden on segmentation algorithm optimization with newer nanopore chemistries, and 3) functions without causing substantial computational overhead. We conclude that CERN provides an effective mechanism to systematically identify and correct the errors in raw nanopore signals before further analysis, which can enable the development of a new class of error correction mechanisms purely designed for raw nanopore signals. CERN is available at https://github.com/STORMgroup/CERN. We also provide the scripts to fully reproduce our results on our GitHub page.
TargetCall: Eliminating the Wasted Computation in Basecalling via Pre-Basecalling Filtering
Dec 09, 2022Basecalling is an essential step in nanopore sequencing analysis where the raw signals of nanopore sequencers are converted into nucleotide sequences, i.e., reads. State-of-the-art basecallers employ complex deep learning models to achieve high basecalling accuracy. This makes basecalling computationally-inefficient and memory-hungry; bottlenecking the entire genome analysis pipeline. However, for many applications, the majority of reads do no match the reference genome of interest (i.e., target reference) and thus are discarded in later steps in the genomics pipeline, wasting the basecalling computation. To overcome this issue, we propose TargetCall, the first fast and widely-applicable pre-basecalling filter to eliminate the wasted computation in basecalling. TargetCall's key idea is to discard reads that will not match the target reference (i.e., off-target reads) prior to basecalling. TargetCall consists of two main components: (1) LightCall, a lightweight neural network basecaller that produces noisy reads; and (2) Similarity Check, which labels each of these noisy reads as on-target or off-target by matching them to the target reference. TargetCall filters out all off-target reads before basecalling; and the highly-accurate but slow basecalling is performed only on the raw signals whose noisy reads are labeled as on-target. Our thorough experimental evaluations using both real and simulated data show that TargetCall 1) improves the end-to-end basecalling performance of the state-of-the-art basecaller by 3.31x while maintaining high (98.88%) sensitivity in keeping on-target reads, 2) maintains high accuracy in downstream analysis, 3) precisely filters out up to 94.71% of off-target reads, and 4) achieves better performance, sensitivity, and generality compared to prior works. We freely open-source TargetCall at https://github.com/CMU-SAFARI/TargetCall.
ApHMM: Accelerating Profile Hidden Markov Models for Fast and Energy-Efficient Genome Analysis
Jul 20, 2022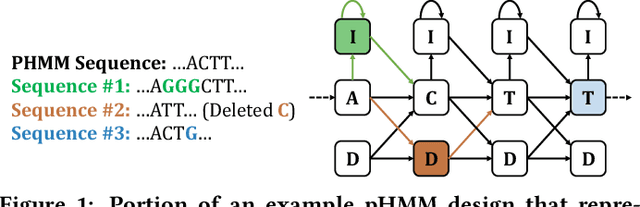
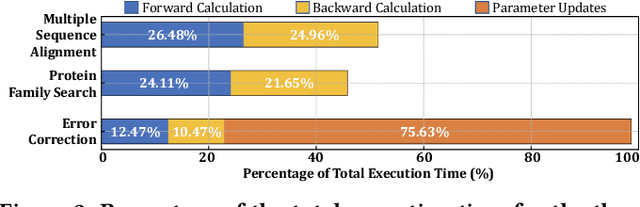
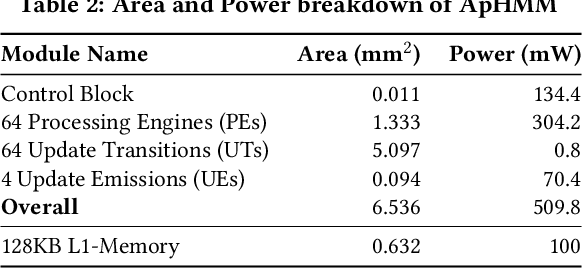
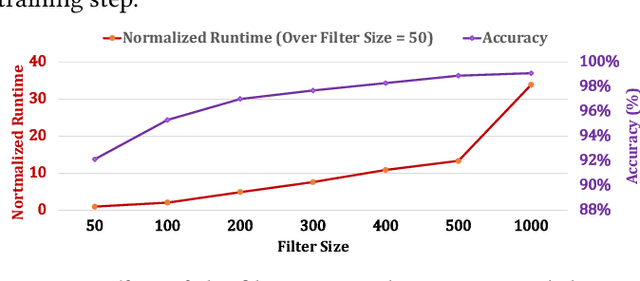
Profile hidden Markov models (pHMMs) are widely used in many bioinformatics applications to accurately identify similarities between biological sequences (e.g., DNA or protein sequences). PHMMs use a commonly-adopted and highly-accurate method, called the Baum-Welch algorithm, to calculate these similarities. However, the Baum-Welch algorithm is computationally expensive, and existing works provide either software- or hardware-only solutions for a fixed pHMM design. When we analyze the state-of-the-art works, we find that there is a pressing need for a flexible, high-performant, and energy-efficient hardware-software co-design to efficiently and effectively solve all the major inefficiencies in the Baum-Welch algorithm for pHMMs. We propose ApHMM, the first flexible acceleration framework that can significantly reduce computational and energy overheads of the Baum-Welch algorithm for pHMMs. ApHMM leverages hardware-software co-design to solve the major inefficiencies in the Baum-Welch algorithm by 1) designing a flexible hardware to support different pHMMs designs, 2) exploiting the predictable data dependency pattern in an on-chip memory with memoization techniques, 3) quickly eliminating negligible computations with a hardware-based filter, and 4) minimizing the redundant computations. We implement our 1) hardware-software optimizations on a specialized hardware and 2) software optimizations for GPUs to provide the first flexible Baum-Welch accelerator for pHMMs. ApHMM provides significant speedups of 15.55x-260.03x, 1.83x-5.34x, and 27.97x compared to CPU, GPU, and FPGA implementations of the Baum-Welch algorithm, respectively. ApHMM outperforms the state-of-the-art CPU implementations of three important bioinformatics applications, 1) error correction, 2) protein family search, and 3) multiple sequence alignment, by 1.29x-59.94x, 1.03x-1.75x, and 1.03x-1.95x, respectively.