 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVariational Encoding of Complex Dynamics
Dec 02, 2017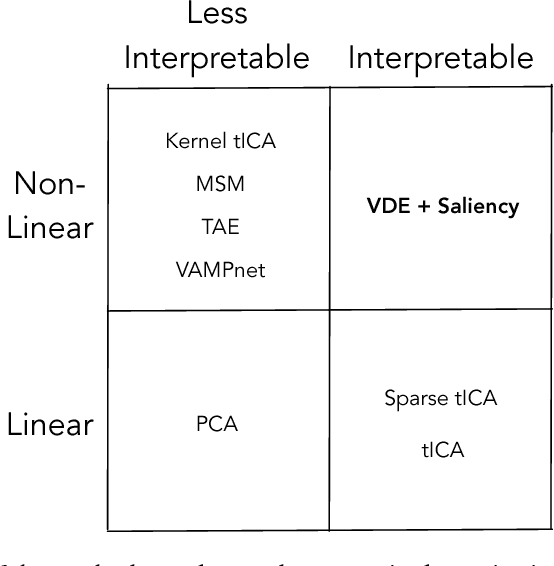
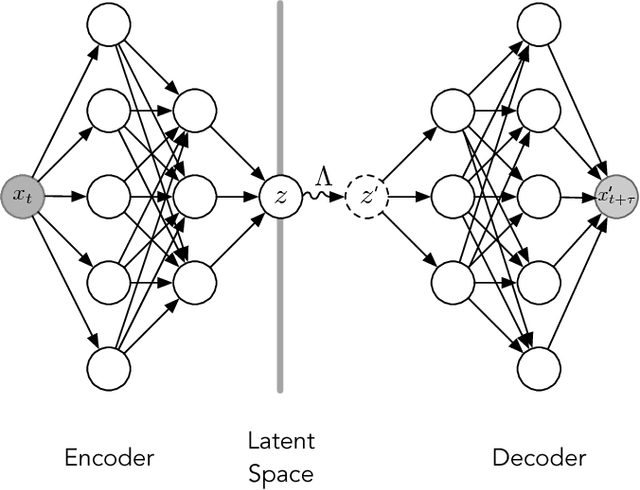
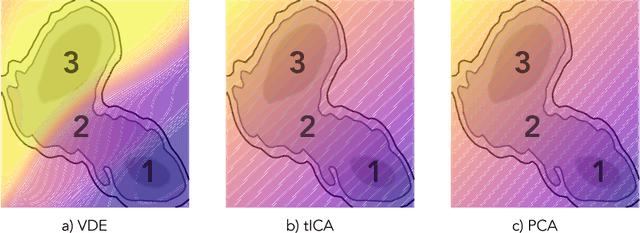
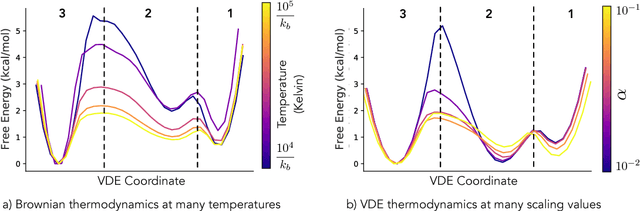
Often the analysis of time-dependent chemical and biophysical systems produces high-dimensional time-series data for which it can be difficult to interpret which individual features are most salient. While recent work from our group and others has demonstrated the utility of time-lagged co-variate models to study such systems, linearity assumptions can limit the compression of inherently nonlinear dynamics into just a few characteristic components. Recent work in the field of deep learning has led to the development of variational autoencoders (VAE), which are able to compress complex datasets into simpler manifolds. We present the use of a time-lagged VAE, or variational dynamics encoder (VDE), to reduce complex, nonlinear processes to a single embedding with high fidelity to the underlying dynamics. We demonstrate how the VDE is able to capture nontrivial dynamics in a variety of examples, including Brownian dynamics and atomistic protein folding. Additionally, we demonstrate a method for analyzing the VDE model, inspired by saliency mapping, to determine what features are selected by the VDE model to describe dynamics. The VDE presents an important step in applying techniques from deep learning to more accurately model and interpret complex biophysics.
* Fixed typos and added references