 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Systematic Approach to Featurization for Cancer Drug Sensitivity Predictions with Deep Learning
May 04, 2020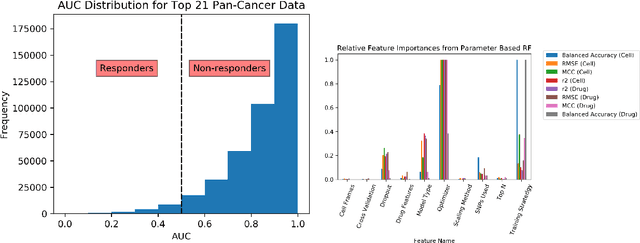
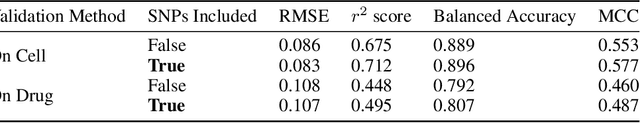
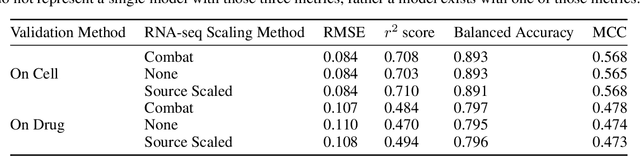
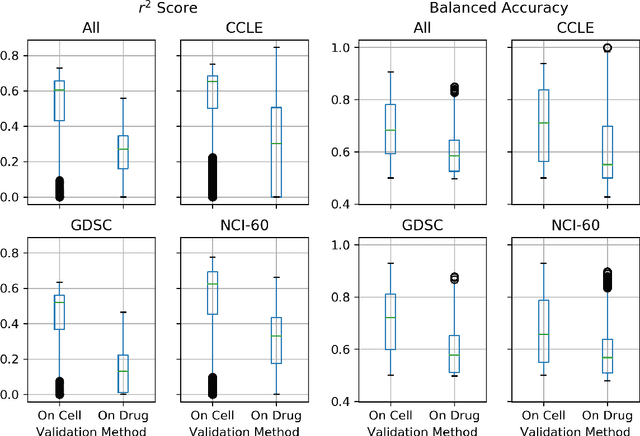
By combining various cancer cell line (CCL) drug screening panels, the size of the data has grown significantly to begin understanding how advances in deep learning can advance drug response predictions. In this paper we train >35,000 neural network models, sweeping over common featurization techniques. We found the RNA-seq to be highly redundant and informative even with subsets larger than 128 features. We found the inclusion of single nucleotide polymorphisms (SNPs) coded as count matrices improved model performance significantly, and no substantial difference in model performance with respect to molecular featurization between the common open source MOrdred descriptors and Dragon7 descriptors. Alongside this analysis, we outline data integration between CCL screening datasets and present evidence that new metrics and imbalanced data techniques, as well as advances in data standardization, need to be developed.